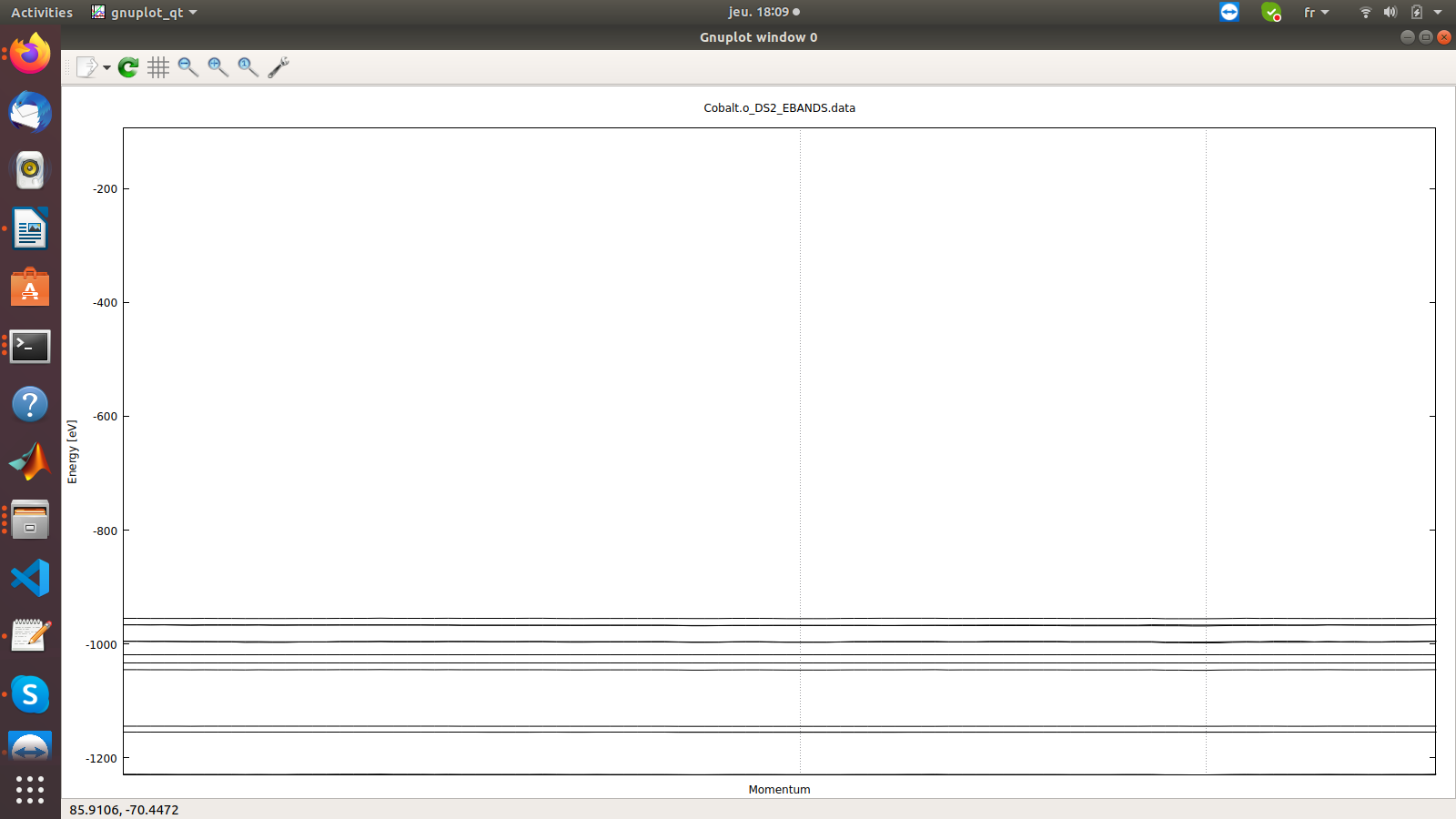
Dear all;,
I tried to make the band structure of monolayer of Cobalt, but what I get is just flat band ,
I attached the input file and the band structure I got ,
I will be grateful for any feedback or remaks
Thank you very much for your comprehnsion
with nice regards
Crystalline Cobalt 13/06/2020
Computation of the band structure.
First, a SCF density computation, then a non-SCF band structure calculation.
ndtset 2
#Dataset 1 : usual self-consistent calculation
kptopt1 3 # Option for the automatic generation of k points,
taking into account the full symmetry
#ngkpt1 4 4 1
prtden1 1 # Print the density, for use by dataset 2
toldfe1 1.0d-12 # This value is way too large for most realistic studies of materials
prtdos2 2
#prtdos1 2
ngkpt1 12 12 1
#Dataset 2 : the band structure
iscf2 -2
getden2 -1
kptopt2 -3 # nb of segments Only 2 segment: G->K and K->M to compare with VASP results
nband2 9
ndivk2 100 70 37 # 10, 12 and 17 divisions of the 2 segments, delimited
by 3 points.
kptbounds 0 0 0 # Gamma
2/3 -1/3 0 # K
1/2 0 0 # M
0 0 0 # Gamma
tolwfr2 1.0d-12
enunit 1 # Will output the eigenenergies in eV
#Definition of the unit cell
#acell 4.737732368 4.737732368 7.690240466
#angdeg 90 90 120
acell 4.737732368 4.737732368 7.690240466
rprim 0.8660254038E+00 5.0000000000E-01 0.0000000000E+00
-0.8660254038E+00 5.0000000000E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 1.0000000000E+00
pseudos “Co.GGA_PBE-JTH”
#Definition of the atom types
ntypat 1 # There is one types of atoms
znucl 27 # The keyword “znucl” refers to the atomic number of the
possible type(s) of atom. The pseudopotential(s)
mentioned in the “files” file must correspond
to the type(s) of atom. Here, the only type is Silicon.
#Definition of the atoms
natom 6 # There are three atoms
typat 1 1 1 1 1 1 # They are of type 1 Mo,type 2 sulphur .
#xred # This keyword indicate that the location of the atoms
1/3 2/3 1/4
2/3 1/3 3/4
#or
xcart
0. 0. 0. #Bohr
4.7377 0 0. #Bohr
0 0. 7.96024 #Bohr
4.7377 0 7.96024 #Bohr
2.36885 4.102968 0. #Bohr
2.36885 4.102968 7.96024 #Bohr
#Definition of the planewave basis set
ecut 20.0 # Maximal kinetic energy cut-off, in Hartree
#Definition of the SCF procedure
nstep 50 # Maximal number of SCF cycles
diemac 12.0 # Although this is not mandatory, it is worth to
precondition the SCF cycle. The model dielectric
function used as the standard preconditioner
is described in the “dielng” input variable section.
Here, we follow the prescription for bulk silicon.
add to conserve old < 6.7.2 behavior for calculating forces at each SCF step
optforces 1
prtebands 2
prtgeo 2
pawovlp -1
pawecutdg 30