Dear Users,
Can someone please help me to get triqs connected with abinit. I have installed triqs 3.1.1 separately on Ubuntu 20.04. Now, how can I tell abinit about triqs? Do I have to recompile abinit using some commands in the .ac9 file? If so, what should I input in the .ac9 file?
Thanks for your help in advance!
Rajesh
Dear Rajesh,
There are three ways to combine ABINIT with TRIQS:
- There is a way to compile TRIQS within ABINIT. However, with the TRIQS API changing often, this is extremely trickly and will probably work for very specific older versions of TRIQS. This way has not be kept up-to-date too much. Also you are limited by the choices of parameters in the way and it’s not optimal.
- You can use the python interface, which calls TRIQS externally. This is a bit tricky and there are no tutorial at the moment on how to do this. With this way, you will be able to perform full charge-self-consistency and you have to use to ABINIT projected local orbitals computed in ABINIT.
- You can use the Wannier90 interface to produce a hr.dat minimal model. Then, with solid_dmft from the TRIQS package, you can perform DMFT. With this you use the Wannier90 orbitals, but you cannot perform full charge-self-consistency at the moment, only one-shot DFT+DMFT.
I am currently working on making the Wannier90 interface fully charge-self-consistent. I will also provide a tutorial which can be used for the python invocation. What exactly are your needs?
Best,
Olivier
1 Like
Dear Olivier,
Many thanks for your comments. Point number 1 is really tricky as you mentioned. I thought about the solution number 3 where I can use the hr.dat file from wannier90 inside abinit. But, as you said there is problem with full charge-self-consistency calculation.
I wanted to check the DMFT results obtained from abinit itself using dmft_solv==5 and triqs cthyb solver from triqs. Also, I wanted to have orbital projected spectral functions. It seems that from abinit, we get total spectral fucntion depending on how many bands you give for the wannierization and I don’t find any option to extract orbital resolved spectral function. Do you have any clue if such options are available in abinit which I might missed?
Are you working on the point no 3?
Regards,
Rajesh
Dear Rajesh,
I am indeed working on point number 3. As for the spectral function, with ABINIT you should have access to the imaginary-time Green’s function for each orbitals, and thus to the spectral functions for each orbitals. I’m not sure what you mean otherwise.
Best,
Olivier
Dear Olivier,
Which file do you meant? I have just Gtau.dat which I guess for all (Total). In addition, I have files like “xxo_DSx_atom_0x_Gtau_0x”. Then I have “xxo_DSxSelfrotformaxent000x_isppol1_iflavor000x” and “xxo_DSxSelf-omega_iatom000x_isppol1”. Which one should be the needed file, I guess the file with ends with _Gtau_0x.
If this is the _Gtau_0x type file I need to work with, then there are many of them, how would I know which of them are for let say 3d x2-y2 orbital?
Or, is it the folder where I did the Omegamaxent calculation using Gtau.dat?
Many thanks!
Rajesh
Dear Rajesh,
As far as I remember, the first column of Gtau.dat should be the Green’s function in imaginary-time, while then there should be two columns for each orbital of the calculation. Isn’t that right? First is the real part and second the imaginary. There might even be inter-orbital components, I’m not sure. Do you have only one column? What is the system you are looking at?
They other files with Self correspond to the self-energy, either at a specific iteration. Then xxo_DSx_atom_0x_Gtau_0x I’m not sure about this, it might be the Gtau of each iteration instead. Does that match you input file?
Best,
Olivier
Dear Olivier,
You are correct. Those xxo_DSx_atom_0x_Gtau_0x files are indeed related with number of iteration and number of steps. I have total 60 files from 12 steps and 5 iteration.
About Gtau.dat, you are right, I have total 11 columns, 10 for five d-orbitals and 1st one is imaginary time Green function. Now, should I extract two columns for each orbital keeping 1st column common and then perform Omegamaxent? Could you please tell me what is the order of those d-orbitals, is it -2,-1,0,1,2?
Thank you!
Regards,
Rajesh
Dear Rajesh,
1st column should be the value of imaginary-time itself, from 0 to beta. You can tell OmegaMaxEnt which columns to select from the file in the input. As for the order, yes it is -2, -1, 0, 1, 2 in real harmonics.
Best,
Olivier
Dear Olivier,
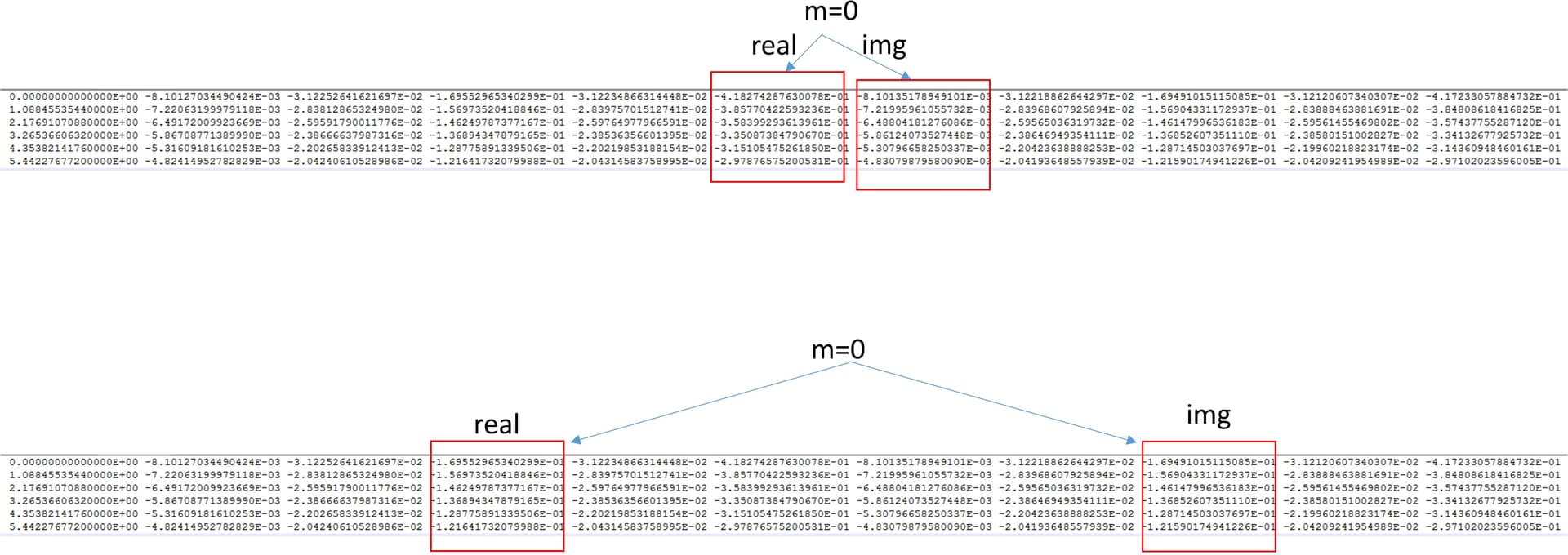
For the notation of orbital (for example m=0) which should be the correct one? (please see the drawings below)
However, it seems bit confusing. I tried with two different approach using the upper notation in the above figure.
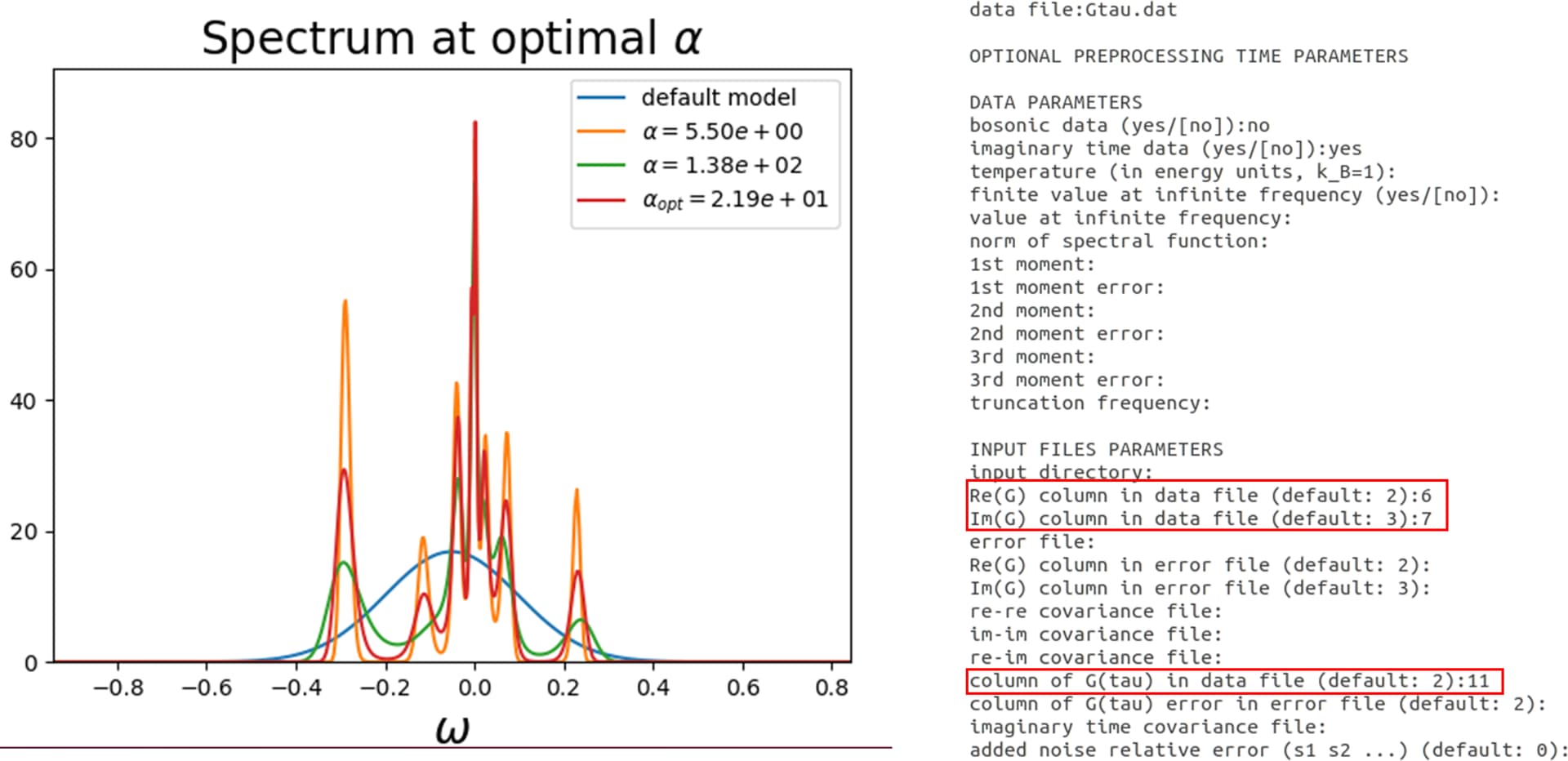
(1) I took the raw Gtau.dat file which contains 11 columns and accordingly the notation, I selected columns 6 and 7 for the z2 orbital.
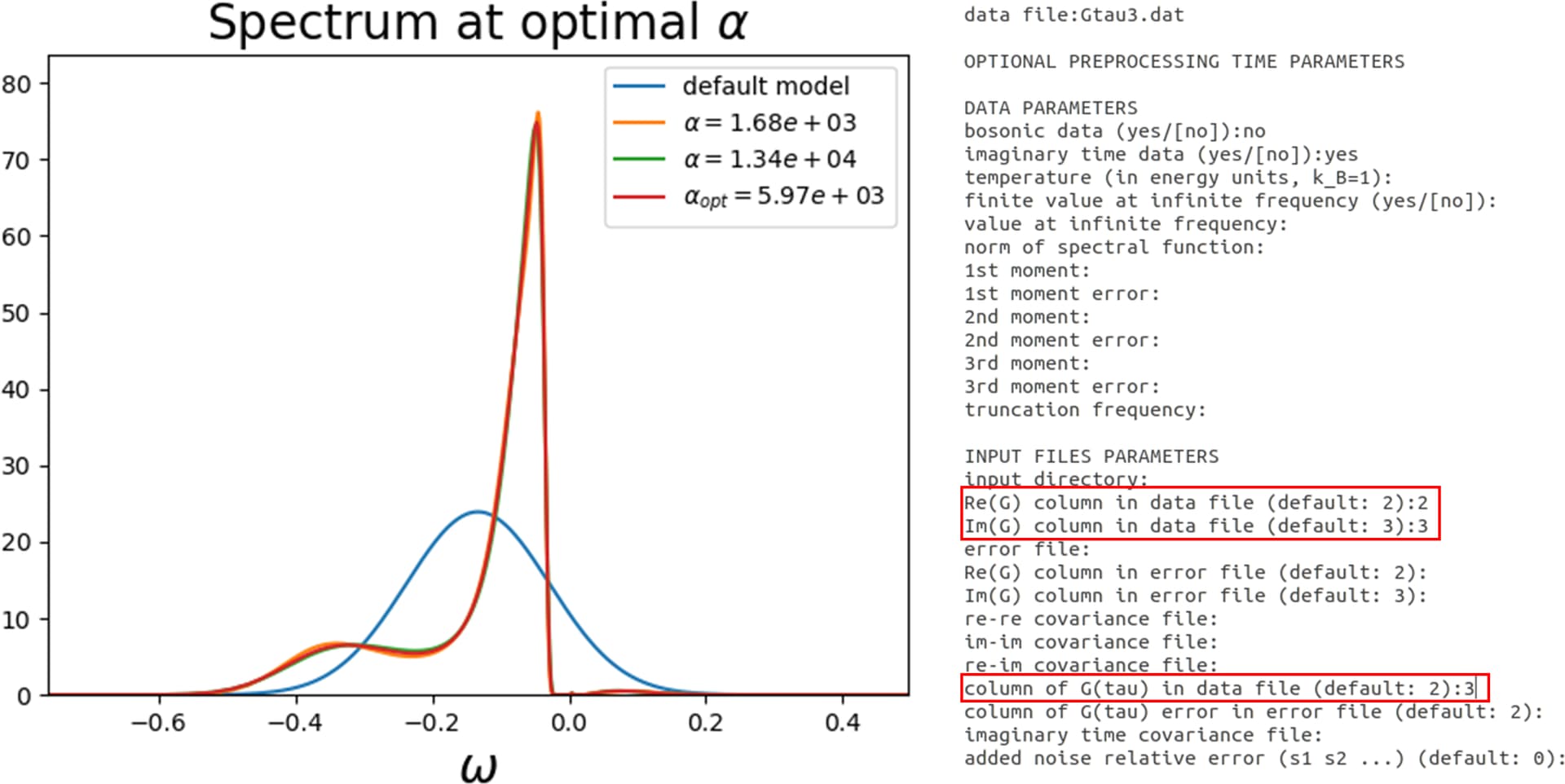
(2) I extracted columns 1, 6 and 7 from raw Gtau.dat file and make a separate file “Gtau3.dat” which contain those columns 1,6 and 7
They give different results. Please see the attachments.
I have also attached the Gtau files here.
Gtau.in (119.4 KB)
Gtau3.in (31.8 KB)
Which one should be correct approach? Do we have to take always both real and imaginary columns for respective orbitals?
Regards,
Rajesh
Dear Rajesh,
Actually, the imaginary-time Green’s function is real. So here you have the up and down components. You can see the first five components are basically repeated for the last five. Also, you seem to have a symmetry between the m=-1 and m=1, which are the yz and zx orbitals. So the correct approach is your second one. You should not use Re(G) and Im(G) in OmegaMaxEnt, this corresponds to imaginary frequencies. You should give 4 or 9 to column of G(tau) instead.
Best,
Olivier
Hi Olivier,
Yes, first five columns are similar to next five columns. Now it is clear. Yes, in PrNiO2 system yz and zx orbitals are seemingly degenerate at least in the paramagnetic state.
I used “column of G(tau) in data file (default: 2):4” for selecting column 4 in the OmegaMaxEnt input file. Is it the correct line to specify the desired column?
However, spectral function using columns 4 and 9 for z2 orbital are slight different and I guess this is because the corresponding values in Gtau.dat file is not exactly same! (see the attachment please)
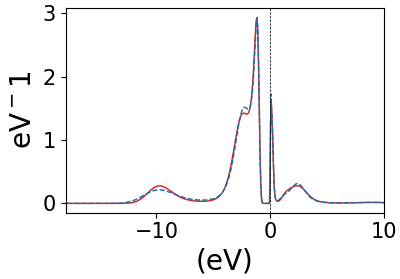
So, what I understood that we have always orbital resolved impurity spectral function from Gtau file and this Gtau file contents the orbitals of the atom on which U has been applied. What about total spectral function, is there any such thing? What I meant is we plot all the spectral functions from 5 d-orbitals together and then sum up to get total spectral function!
Anyway, many thanks for your clarification and it was great help.
Regards,
Rajesh
Dear Rajesh,
Due to quantum Monte Carlo and analytical continuation, it is very hard to have exactly equal spectral functions. Those look great! As for the total spectral function, indeed this is like a DOS, so you can some all the contributions from each orbital to get the total one for the d-shell of Ni in this case.
Best,
Olivier