I mentioned to you in our previous discussion that increasing “dmftqmc_n” beyond 1.d9 gives an error “ERROR in QMC rank 0 : Ctqmc_setSweeps : sweeps is negative or too big”. I have even more different situation here.
First, let me ask you one question: in the DMFT tutorial for k-resolved spectra section, the input tdmft_5.abi has three datasets. Does the dataset 3 take the same dmftqmc_n as in dataset 2 for DMFT? If yes, then I don’t understand the problem I am having.
I tried to calculate the 2nd dataset setting dmftqmc_n=1.d10, and it works (the results are in the previous discussion) but, it throws the above error for data set 3. Therefore, I changed it 1.d09 for dataset 3 and it worked.
A second problem is that with dmftqmc_n=1.d.11, it throws the same error even for dataset 2.
I guess it must be related with memory or mpi problem. Here I attach the log file for the second problem. log-dataset2.in (485.8 KB)
As for dataset 3 of the tutorial, it does not perform QMC. You see, you provide it with a self-energy that is already analytically continued, so it does not run DMFT, it just takes your self-energy, reembeds it to the lattice, and compute the spectral function. QMC is to obtain the self-energy on the imaginary axis, which is done in dataset 2.
As for the error you get, I do not really understand it. Maybe @amadon could enlighten us as to why it is there? There is no a priori reason to limit the number of sweeps. It does not affect the memory, only the walltime. I’m not sure what to tell you about that, apart from the fact that your self-energies look good enough at the moment (based on figure from another post).
Exactly this is what I thought but, just wanted to confirm. This is strange! Reducing sweeps to n9 for dataset 3 it did not through error while, dataset 2 was done using n10.
BTW, does dmft_rslf play a huge role for obtaining self-energy in the dataset 2? I have noticed another strange behavior. I will come back to you after detail investigation.
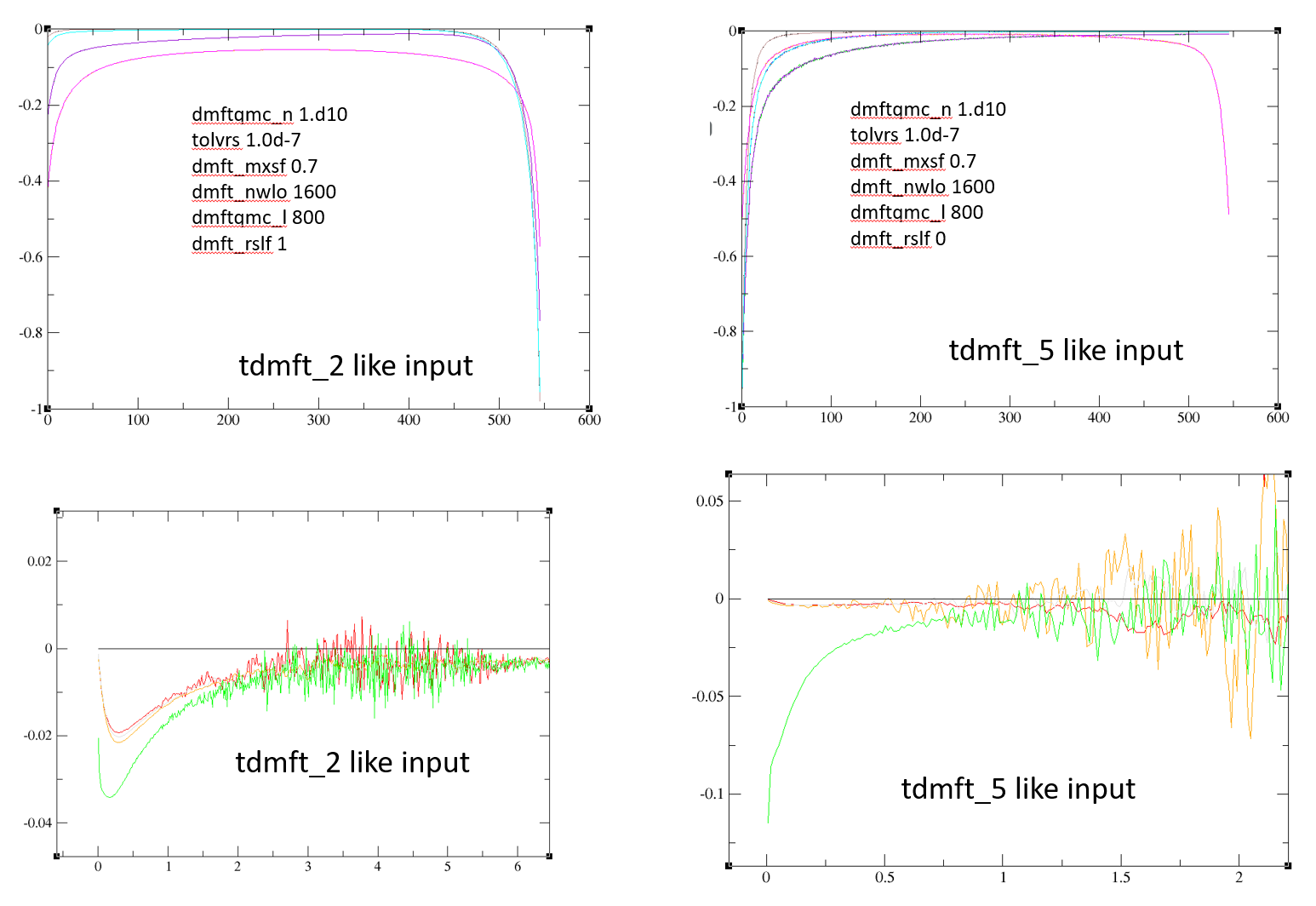
I have compared the two calculations for my case (1) similar input like in tdmft_2.abi and (2) tdmft_5.abi keeping convergence and other parameters same.
In the tutorial, they both intend to perform DMFT in the dataset 2. The difference between two input files which affect the results is “dmft_rslf”. For tdmft_2.abi it is 1 and for tdmft_5.abi it is 0. Also, in tdmft_2.abi we have nline1, nnsclo1==5 which has default values in tdmft_5.abi.
There are few extra dmftctqmc_XX variables in tdmft_5.abi and most of them have default values except dmftctqmc_correl.
I got two different Gtau and self-energy plot from two calculations. Here is the plots. It seems like input file like tdmft_5.abi needs more convergence, but it does not go below 1.d-7 even with higher dmftqmc_l and dmft_nwlo.
What could be the reason to have different results.
I’m not sure what is the difference between the calculations. Clearly, they have different chemical potential. The one on the right is almost a band insulator. You should provide your files, but looking at the tutorial, tdmft_5 has nstep2 1 and dmft_iter 10. I think it might not be converged at all at the DFT level. I would suggest inverting these keywords. I should also probably go through this tutorial and correct it if that is the case. The Green’s function you get should give totally different k-resolved spectral function than the one from the tutorial.
Let me know, if there are problems I will go through it and make the changes,
Best,
Olivier
Those calculations are for PrNiO2 and I prepared two input files like one as tdmft_2 and other as tdmft_5. My feeling is the rslf variable make this difference. If you have time, please check for SrVO3 case: take tdmft_5 input one with dmft_rslf==0 and another calculation with dmft_rslf==1. You can switch nstep2 and dmft_iter. This I already did for PrNiO2 for both calculations and results are like above.
I understand better than. The dmft_rslf parameter only reads the self-energy and is used to restart calculations. It should not affect anything, apart from if there is already a file in the folder. I cannot help you without the files you used. But at this point it becomes interpreting data and performing research, which is not the goal of this forum.
Ideas: if you changed the temperature, the interaction or the number of orbitals, this will definitely change your calculation.
Thanks for your reply. I still do have confusion!
I have performed two calculations on SrVO3 using tdmft_2 and tdmft_5 input files.
What I changed in the input file tdmft_2.abi are: nstep2==30, dmft_iter==1.
For the tdmft_2.abi, I have changed the followings in order to keep the convergence variables same as in tdmft_5.abi: ecut, pawecutdg, ngkpt, dmft_nwlo, dmftqmc_l,dmftqmc_therm, tolvrs and dmft_mxsf.
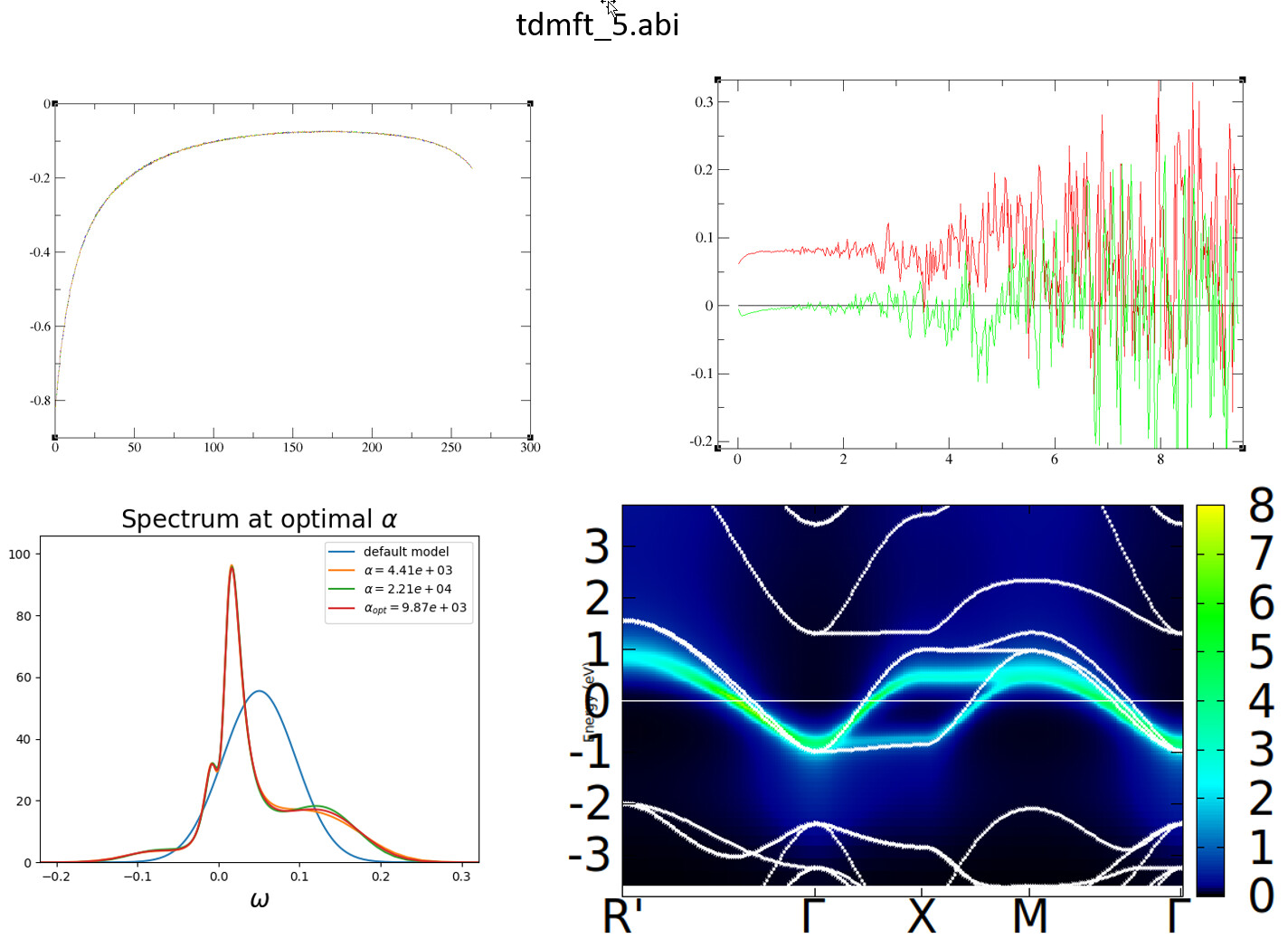
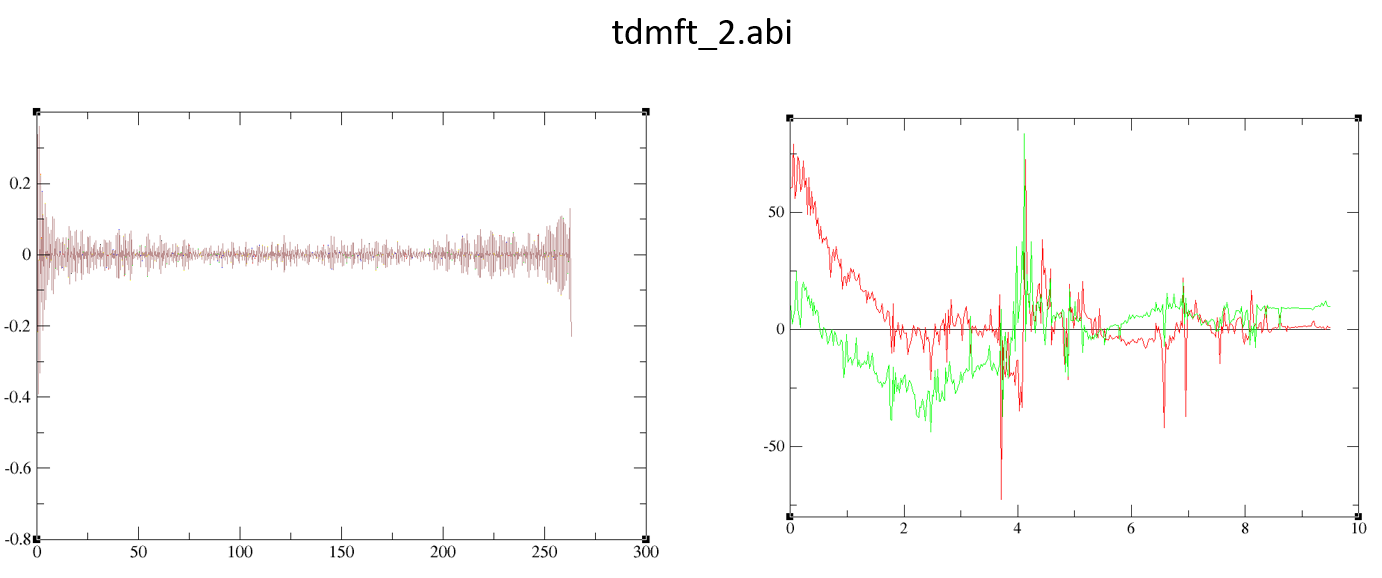
I have attached the both input files and the results. For the k-resolved spectra (tdmft_5.abi) it is very good but, for tdmft_2.abi, results are far away from reality.