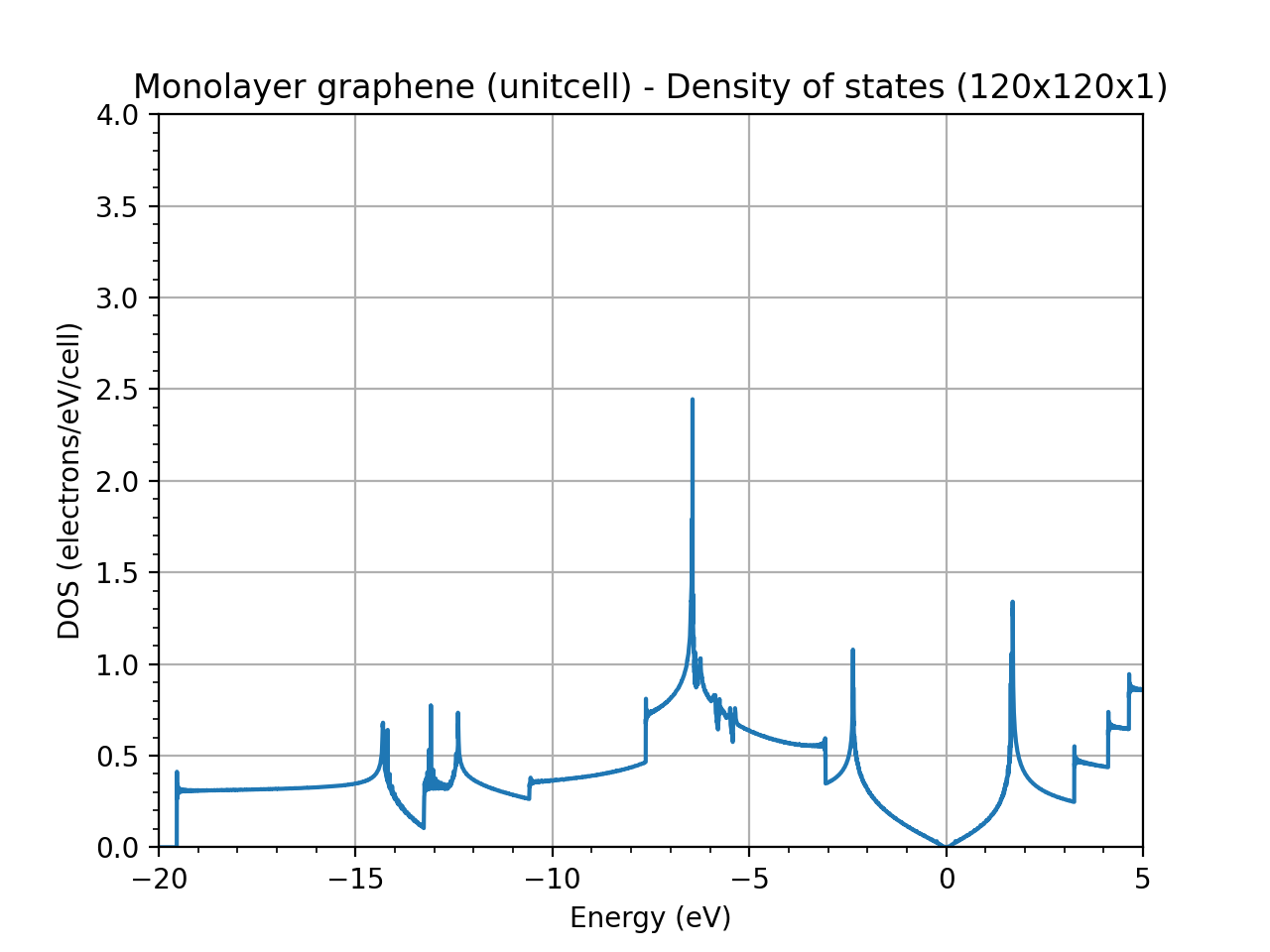
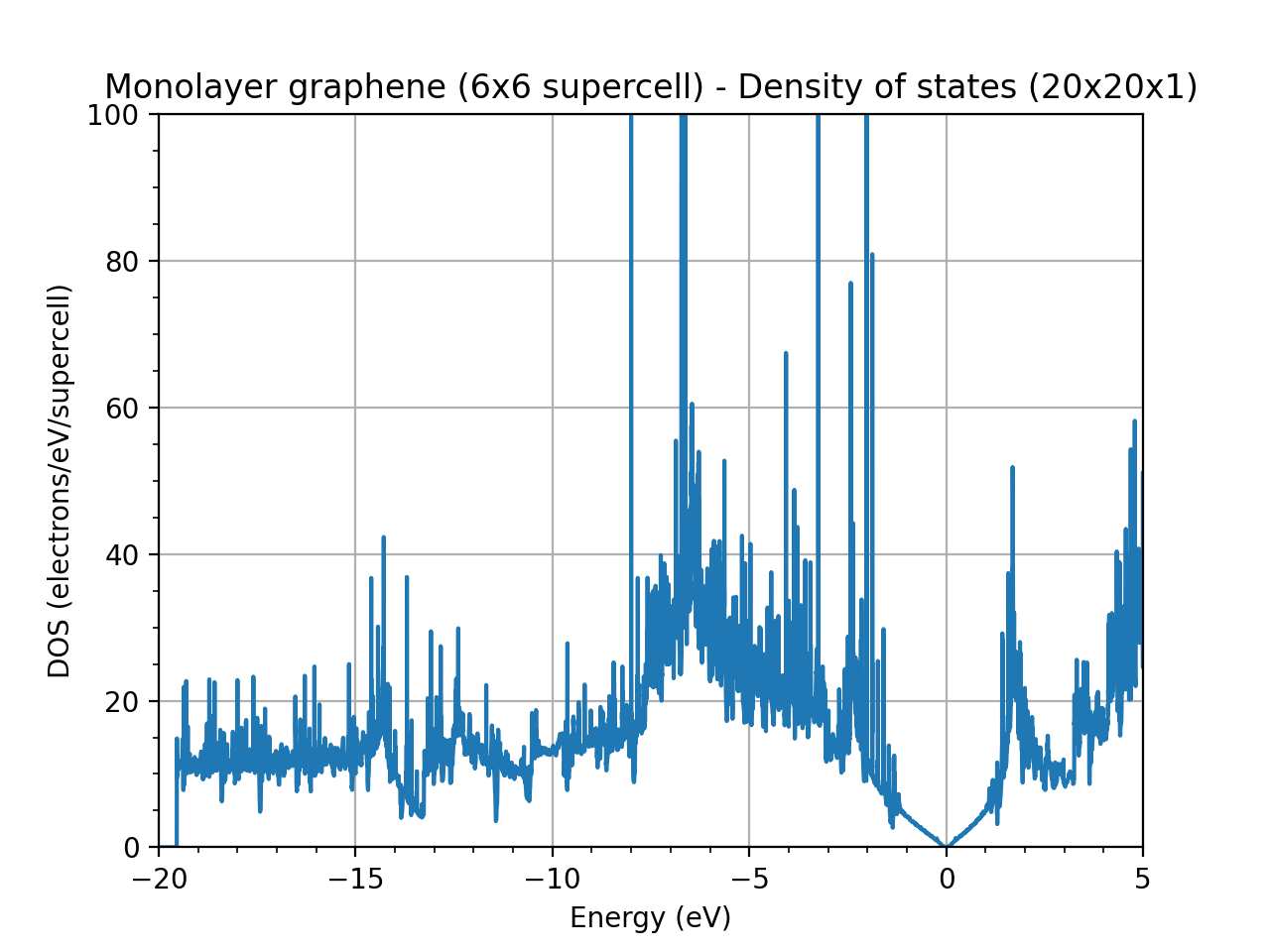
I am doing some DOS calculations for pristine graphene. Since I am working on the functionaliztion of graphene, I am using a supercell for my calculations. However, when I calculate the DOS of graphene using a 6x6 supercell instead of the unitcell, the plot that I have is very noisy. For reference, I used a k-point grid of 120x120x1 in the unitcell case and of 20x20x1 in the supercell case in order to maintain the same resolution. Any ideas on why the DOS is so noisy for the supercell case?
Even if you are respecting the inverse relation between real and reciprocal space, in supercell calculations, reducing the k-point sampling often results in a noisier Density of States (DOS) because you lose resolution in the Brillouin zone integration. In the unit cell, a denser grid of 120 120 1 provides smooth DOS, but in a larger supercell with 20 20 1, you’re effectively integrating over fewer points, which can make the DOS appear noisier.
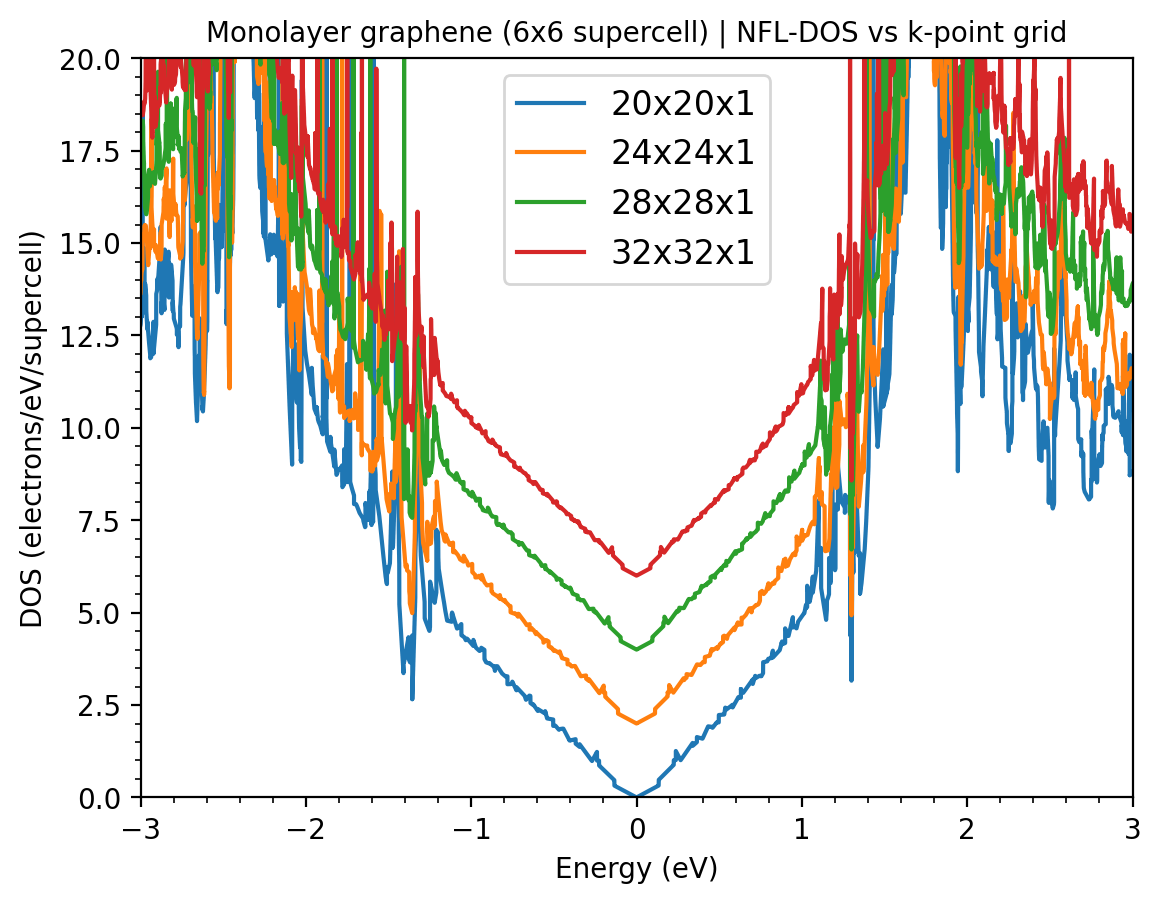
You can try to increase a bit the smearing parameter to 0.002 or 0.003 Hartree or increase ngkpt slightly beyond the inverse relation. ie 24 24 or 30 30 depending on your computational resources.
At least two possible effects here. 1) your positions in the supercell have 10 digits “only” so maybe you are slightly breaking the translational symmetry and this shifts some peaks around.
More probable is 2) the tetrahedron has a hard time with band crossings, and by making a supercell you are producing tons of them. The tetrahedron is done band by band, and interpolates the energies for 1 band, without being able to check if the band character ordering is different at k1, k2, k3, k4. If you have a crossing in the middle of the tetrahedron, the bands look flatter than they should, and you get spikes (similar to this example of skw interpolation). If you have a fully degenerate tetrahedron (e1=e2…) then its DOS is replaced by a very thin gaussian, which adds more spikes.
Try with Gaussian integration and converge the width: For the tetrahedron case it does ignore the smearing.