Dear All ABINIT Users,
I hope this message finds you well.
My name is Diva, and I am a new user of ABINIT. Please kindly excuse me if my question seems basic or has been addressed before.
I am currently working on a project involving a two-dimensional material, Fe₂Si, with interstitial doping using Chromium (Cr). The primary objective is to investigate its electronic structure and magnetic properties using Density Functional Theory (DFT) with ABINIT. While analyzing the electronic structure, I noticed some discrepancies in the results. Specifically, I obtained different DOS and PDOS plots depending on the value of the prtdos variable — using prtdos 2 for DOS, and prtdos 3 for PDOS. I am trying to understand why this happens.
In one of the tutorials (for crystalline silicon), I observed that the ngkpt value used for the DOS calculation (dataset 2) was larger than the one used for the SCF calculation (dataset 1). I suspect that a similar issue with the k-point grid might be affecting my own results, but I am not entirely certain.
I would be very grateful if you could help me understand whether this difference is expected, and if there is anything I may be missing in my setup.
Thank you very much for your time and assistance. I sincerely appreciate your support.
Warm regards,
Diva
Dear Diva,
Welcome to the ABINIT community!
Could you be more precise about the “differences” that you see between the results?
As you probably know, the total density of states (DOS), printed with prtdos 2 sums over all states and doesn’t distinguish between atoms or orbitals as done with prtdos 3. So they require some processing to be comparable. Are you talking about these differences?
As you correctly noticed from the tutorial, it’s a common and recommended practice to use a denser k-point grid for the DOS calculation than for the initial SCF step. This ensures better energy resolution in the Brillouin zone integration and smoother, more accurate DOS/PDOS plots.
One caution, just in case: if you change the k-point grid between the SCF and NSCF calculations (i.e., use a denser k-point grid for the DOS/PDOS), you cannot directly reuse the WFK file from the SCF run. This is because the wavefunctions are calculated on the specific k-point grid, and changing the grid will invalidate the wavefunction from the previous calculation. In this case, you’ll need to perform a new NSCF calculation to generate the wavefunctions on the new k-point grid.
Best regards,
Fernando
Dear Fernando,
Thank you very much for the warm welcome and your detailed explanation!
Yes, the differences I observed are indeed related to the Fermi energy values obtained when using prtdos 2 and prtdos 3. Specifically, I noticed a shift in the Fermi level between the two cases, which affects the alignment of the DOS plots. I understand that prtdos 2 sums over all states without resolving atomic or orbital contributions, while prtdos 3 provides projected DOS, and that some processing is needed to make them comparable.
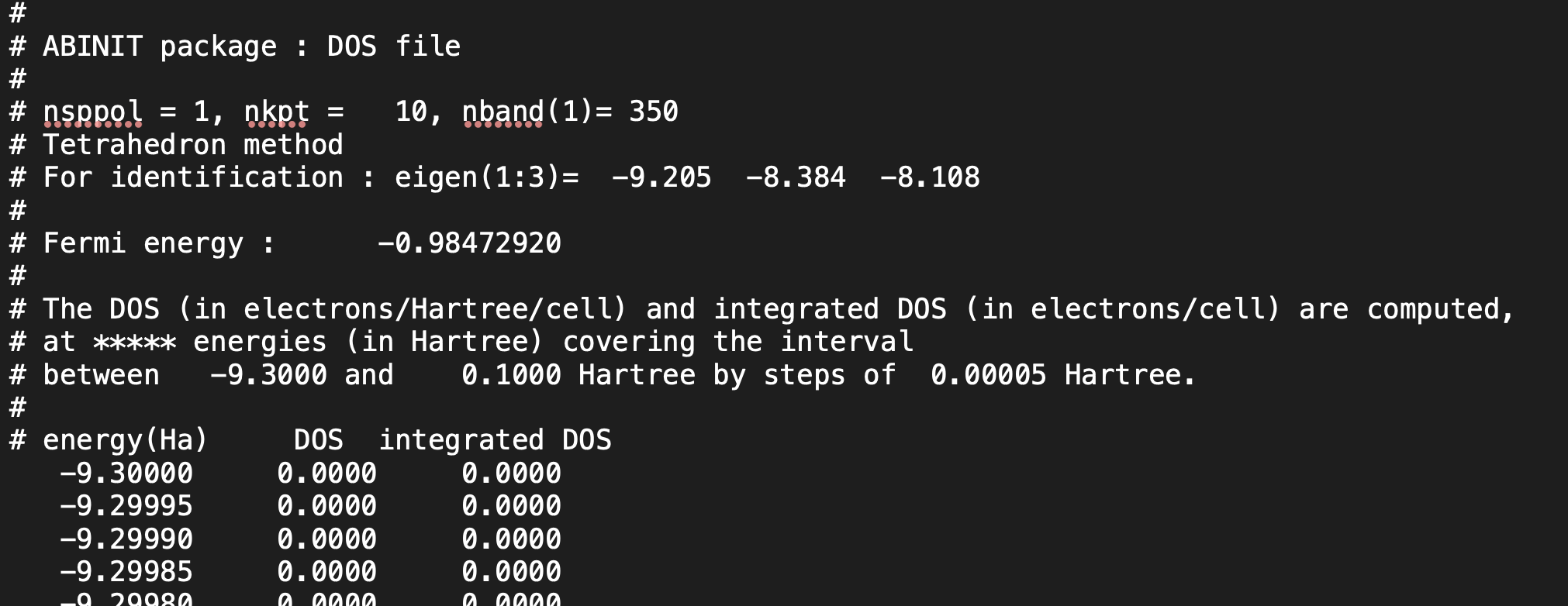
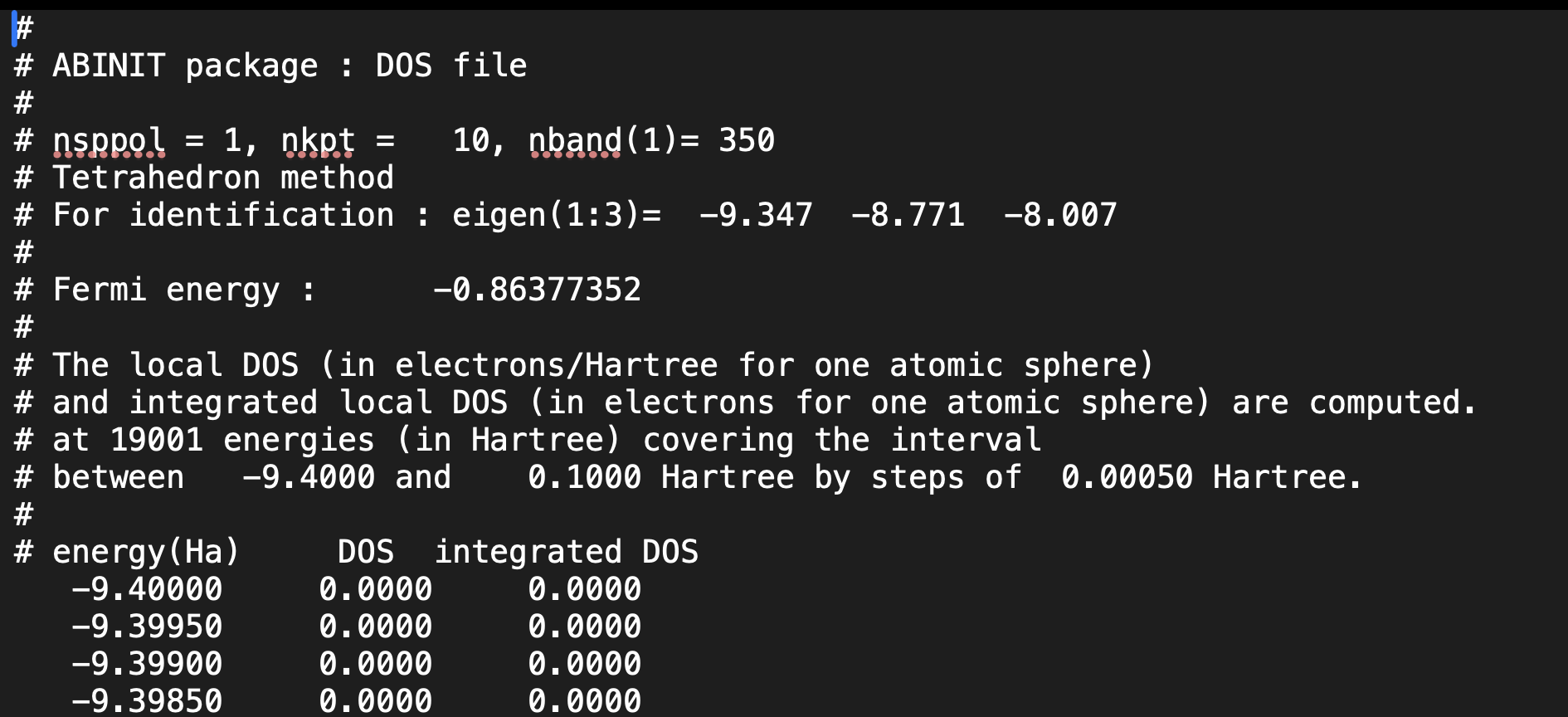
To clarify my concern further, I have attached the input, output, and DOS files for both calculations.
However, even after following these steps, the Fermi energy reported in the *.DOS and *.pdos files seems to differ slightly. Could this be due to the way ABINIT calculates the DOS for prtdos 3 (i.e., orbital projection), or is there an additional normalization step that could affect the Fermi level?
I would greatly appreciate your insight into this, as I want to make sure I am interpreting the data correctly.
Best regards,
Diva
PRTDOS 2
Cr-B_prtdos2.abi (2.9 KB)
Cr-B.abo (227.1 KB)
PRTDOS 3
Cr-B_prtdos3.abi (2.9 KB)
Cr-B.abo (186.8 KB)
Dear Diva,
I’d recommend refining the k-point mesh, especially for DOS calculations, to improve the resolution of the Fermi level and the overall DOS features. A finer grid would provide a more accurate result, though keep in mind that if the grid is too fine, it can lead to an unnecessarily large number of bands being included in the calculation.
I would also recommend you reducing the number of bands used by employing a more optimal value to avoid overkill. Now you are using 350 bands and many of them don’t play any role at all while they can cause potential issues.
Best regards,
Fernando
Dear Fernando,
Thank you very much for your suggestions. I am currently trying to improve the k-point mesh, especially for the partial density of states calculations, to achieve more accurate results.
I would also like to ask whether the parameters natsph and iatsph need to be explicitly declared in this type of calculation? I want to make sure my settings are correct.
Thanks in advance for your help.
Best regards,
Diva
Dear Diva,
These cariables are usefull if you are interested in projecting onto specific atoms or orbitals. These parameters are typically used when performing site- or orbital-resolved projections, where you want to analyze the contribution from particular atoms or angular momentum channels.
If you are interested in the full PDOS you don’t need to explicitly set them.
Best regards,
Fernando
Dear Fernando,
Thank you very much for your explanation. That clarified the usage of the variables.
I would like to inform you that the results from prtdos 2 and prtdos 3 are consistent in my case.
However, I have one question related to magnetic calculations. In the ABINIT tutorial below,
https://docs.abinit.org/tutorial/spin/
the spin DOS plot for magnetic systems is generated using prtdos 1. If I want to perform a similar magnetic calculation but using prtdos 3, is it possible to directly replace prtdos 1 with prtdos 3 in the input file from the tutorial? Will it still give a correct spin-resolved DOS?
Thank you for your time and assistance.
Best regards,
Diva