Hello Abinit community,
I tried to calculate the electronic band structure following Abinit tutorial. I set kptbounds to
0.5 0.25 0.75
0.5 0.5 0.5
0.0 0.0 0.0
0.5 0.0 0.5
0.75 0.25 0.5
0.75 0.375 0.375
but when I tried to visualize it using abiopen.py script, it’s gave another circuit (M gamma L Gamma) . As I need to keep the same circuit in order to compare two band structures. Can you tell me please what the problem is ??
Thank you in advance.
Regards
Hello,
we need much more information here to be useful: input, crystal structure, perhaps machine and installation of abi{nit,py}.
I suspect that abipy is recognizing the symmetry on its own (perhaps wrongly) as hexagonal and imposing those k point paths.
Abinit should listen to your kptbounds, and generate an EIG file correspondingly, which can be parsed and plotted. There is a text version and I think still an .agr xmgrace format file, which can be plotted easily.
The default abipy run may be using the SCF ground state files, and not the nscf ones, and plotting its own path.
Hello,
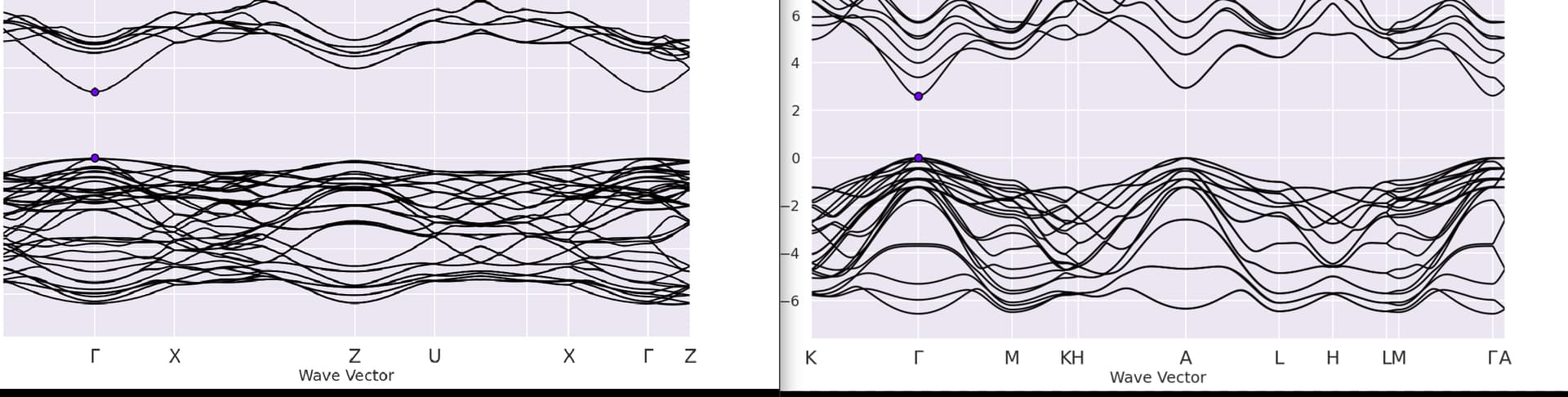
thank you so much for your reply. I redid the calculations, the structures are wurtzites with two different atomic configurations, so I used the kptpounds of an hexagonal structures as mentionned in abinit tutorial:
kptpounds
1/3 1/3 0 0 0 0 1/2 0 0 1/3 1/3 0 1/3 1/3 1/2 0 0 1/2 1/2 0 1/2 1/3 1/3 1/2 1/2 0 1/2 1/2 0 0 0 0 0 0 0 1/2
then I visualized the band structure stored in the GSR.nc file, using the py script and the command line:
abiopen.py band_DS2_GSR.nc --expose sns=talk
I have always got different circuit ( see the attached image) although I declared the same kptpounds.
I add that I have installed the Python 3.11.3 version on *64bit ubuntu desktop, but the calculations were done on a cluster, with abinit 8.8.4.
Best regards,