I made an electron-phonon calculation with the two available codes (legacy, new) starting from the same phonon calculation and ground state on gold with a norm-conserving pseudopotential and I found different linewidths and spectral functions. How is that possible?
useful check, thanks for flagging it. The workflows are not identical in the 2 methods, so I would not expect them to yield exactly the same results, but hopefully close. In particular: legacy does a FT interpolation of the matrix elements, which in general is not a great thing (can go negative if the interpolation is not smooth enough). The new method should be better, FT interpolating \delta V_{SCF}, then taking explicit matrix elements on the k points you need for the Fermi Surface.
I think the eph file you provide does not actually do any q point interpolation on \delta V_{SCF}. You are missing several key input variables, especially eph_ngqpt_fine which determines the q grid for potential interpolation.
There is a fallback to interpolate the rest of the way (if the q path is finer than eph_ngqpt_fine=ddb_ngqpt= 6 6 6 in your case) using the old method of FT interpolation on the matrix elements. If this is what you were aiming to do, to compare with legacy, it would have been useful to explain in your post. It looks like the new matrix element interpolator is having a cow and producing all of these oscillations due to FT aliasing.
Could you try the “normal new” interpolation method with the potential as well to compare?
A2F differences may also be linked to the integration method, between gaussian, tetrahedron etc… The new one is probably integrated only on the 6 6 6 q grid, but the 30 30 30 k grid. See ph_ngqpt. The low energy part may be problematic if it’s not parabolic (Debye model like) for the integration of superconducting quantities.
What is the difference between NC and PAW? All of your inputs are a bit mixed, with NC psp8 files, and pawecutdg defined as well (normally should not be).
So: there may still be an issue, e.g. if the FT interpolation of the matrix elements in the new workflow does not have the proper weights or bravais lattice, but this is not the normal intended use of the new code. Please try the suggestions above and we will see
Actually what I have done is using eph_ngqpt_fine 30 30 30 in input and plotting output file “Au_NOINTP_A2FW” in order to exclude issues arising from interpolation while comparing with legacy. Sorry for not telling in the post.
I didn’t know the code was interpolating anyway according to the ph_qpath following the old method. My aim was indeed to compare with legacy before interpolation.
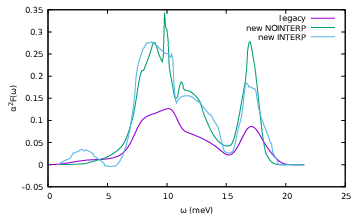
I made a new plot with both output functions. The “normal” new interpolation method yields even worse results compared to legacy but that’s probably just a convergence issue.
I am using tetrahedra everywhere. As far as I understand, A2F differences should not depend on this as long as the integration grid is the same.
The PAW calculation yields approximately the same result as the NC one with the legacy code, but that’s not pertinent to the issue. Forget about it.
Thanks again and let me know if you have other suggestions please.
Also, I really believe the old version is not using the tetrahedron method, even if you gave it the corresponding input variables… The curve is much too smooth. Another sign is the decay rate near omega=0, which should be quadratic in the real physical function, but is a sum of gaussians in the gaussian smearing case, and a weird splined thing in the tetrahedron case.
The new interpolated case is certainly evil. Just to remind me: it does 1) perturbed potential interpolation (which should be fine) or 2) matrix element interpolation (which is worse and can give negative a2F) ?
The new code should be doing perturbed potential interpolation while the legacy code does matrix element interpolation via anaddb according to the documentation.