Hello all,
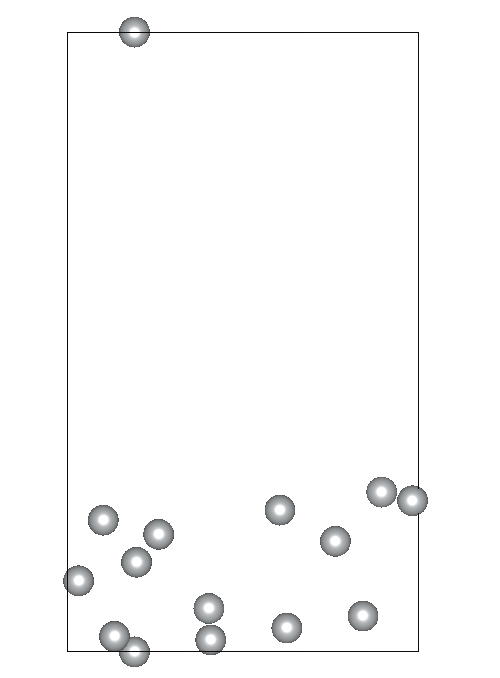
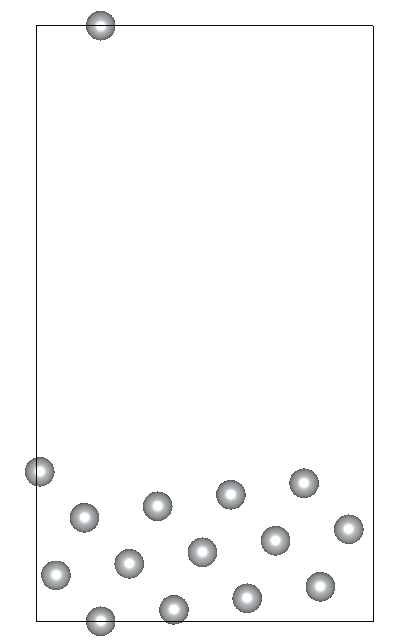
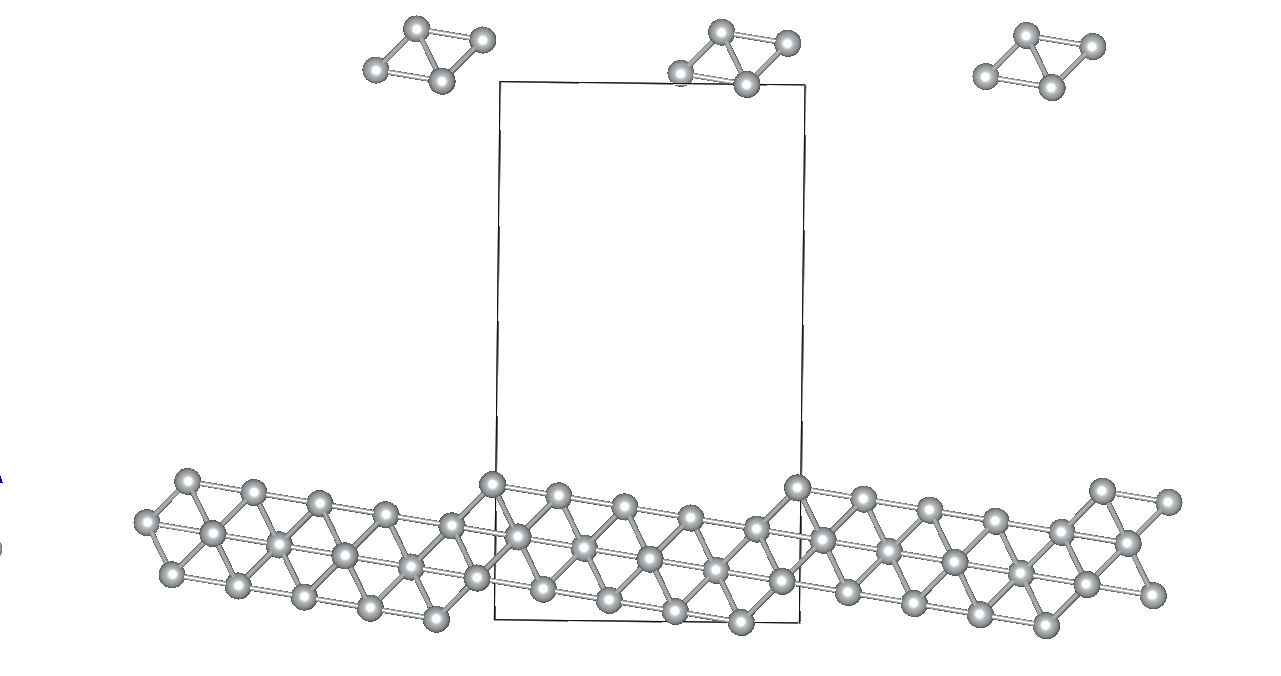
I am working on relaxing a Ni(119) slab composed of three atomic layers and a vacuum slab (15 angstroms). I’ve constrained the positions of the bottom atomic layer while allowing the two top layers to relax. Despite my efforts, the Ni(119) surface becomes significantly deformed during optimization.
To initiate my tests (quick test on convergence), I’ve set a low cut-off energy (5 Ha), a minimal number of k-points, and relaxed convergence criteria. My aim is to ensure everything is configured correctly before proceeding with more intensive computations. Below are the critical parameters from my input file:
##############################################
SECTION: basic
##############################################
ecut 5
ixc 11
ngkpt 1 1 1
shiftk 0.5 0.5 0.5
nshiftk 1
kptopt 1
nsppol 1
nband 550
occopt 3
#tolmxde 0.0005
#toldfe 0.0001
tolmxf 5.0d-3
ionmov 2
iscf 7
npulayit 5
chkprim 0
optforces 2 # Enable force optimizations
timopt 1 # Enable time optimization
prteig 1 # Print eigenvalues
prtwf 1 # Request ABINIT to write the wavefunction file
prtden 1 # Print electron density
ntime 30
nstep 30
toldff 5.0d-4
optcell 0
#SCF preconditioner
iprcel 45
Restarting from the last configuration in the HIST.nc file
#restartxf -3
Specify the path to the DEN file directly for initializing the electronic density
#getden_filepath “Niopt_119out/out_TIM4_DEN”
##############################################
SECTION: files
##############################################
indata_prefix “indata/in”
tmpdata_prefix “tmpdata/tmp”
outdata_prefix “outdata/out”
pseudos “…/Ni119/Ni.psp8”
##############################################
SECTION: gstate
##############################################
chksymbreak 0
nspinor 1
nspden 1
charge 0.0
tsmear 0.01 Ha
chksymtnons 0
##############################################
GeoConstraints
##############################################
natfix 16
iatfix 9 23 37 51 5 19 33 47 11 25 39 53 7 21 35 49
##############################################
STRUCTURE
##############################################
natom 56
ntypat 1
typat
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1
znucl 28
xred
0.0073300000 0.0586000000 0.0772500000
0.0013000000 0.0104100000 0.2510600000
0.0615400000 0.4923400000 0.1158800000
0.1157600000 0.9260700000 0.1545000000
0.1760000000 0.4080000000 0.0193100000
0.1699800000 0.3598100000 0.1931300000
0.2302200000 0.8417300000 0.0579400000
0.2241900000 0.7935400000 0.2317500000
0.1488900000 0.1911300000 0.0000000000
0.1428700000 0.1429400000 0.1738100000
0.2031100000 0.6248700000 0.0386300000
0.1970800000 0.5766700000 0.2124400000
0.0344300000 0.2754700000 0.0965600000
0.0886500000 0.7092000000 0.1351900000
0.2573300000 0.0586000000 0.0772500000
0.2513000000 0.0104100000 0.2510600000
0.3115400000 0.4923400000 0.1158800000
0.3657600000 0.9260700000 0.1545000000
0.4260000000 0.4080000000 0.0193100000
0.4199800000 0.3598100000 0.1931300000
0.4802200000 0.8417300000 0.0579400000
0.4741900000 0.7935400000 0.2317500000
0.3988900000 0.1911300000 0.0000000000
0.3928700000 0.1429400000 0.1738100000
0.4531100000 0.6248700000 0.0386300000
0.4470800000 0.5766700000 0.2124400000
0.2844300000 0.2754700000 0.0965600000
0.3386500000 0.7092000000 0.1351900000
0.5073300000 0.0586000000 0.0772500000
0.5013000000 0.0104100000 0.2510600000
0.5615400000 0.4923400000 0.1158800000
0.6157600000 0.9260700000 0.1545000000
0.6760000000 0.4080000000 0.0193100000
0.6699800000 0.3598100000 0.1931300000
0.7302200000 0.8417300000 0.0579400000
0.7241900000 0.7935400000 0.2317500000
0.6488900000 0.1911300000 0.0000000000
0.6428700000 0.1429400000 0.1738100000
0.7031100000 0.6248700000 0.0386300000
0.6970800000 0.5766700000 0.2124400000
0.5344300000 0.2754700000 0.0965600000
0.5886500000 0.7092000000 0.1351900000
0.7573300000 0.0586000000 0.0772500000
0.7513000000 0.0104100000 0.2510600000
0.8115400000 0.4923400000 0.1158800000
0.8657600000 0.9260700000 0.1545000000
0.9260000000 0.4080000000 0.0193100000
0.9199800000 0.3598100000 0.1931300000
0.9802200000 0.8417300000 0.0579400000
0.9741900000 0.7935400000 0.2317500000
0.8988900000 0.1911300000 0.0000000000
0.8928700000 0.1429400000 0.1738100000
0.9531100000 0.6248700000 0.0386300000
0.9470800000 0.5766700000 0.2124400000
0.7844300000 0.2754700000 0.0965600000
0.8386500000 0.7092000000 0.1351900000
acell 1.0 1.0 1.0
rprim
18.8352782294 0.0000000000 0.0000000000
-2.3544352899 21.4497631255 0.0000000000
0.0000000000 0.0000000000 37.8481907145
Given these settings, the surface deformation I’m observing is unexpected and suggests potential issues with my relaxation strategy or the parameters chosen. By the way the constraints applied to the bottom layer are correctly implemented.. I am particularly concerned whether:
- The low
ecutvalue and sparse k-point grid could be contributing to the deformation. - Any other parameters in my input file could be optimized for better handling of slab relaxation without compromising the integrity of the surface.
I have attached my full input file for reference. Could anyone provide insights into these issues or suggest adjustments to improve the relaxation results? Any advice on best practices for slab relaxation in ABINIT would also be greatly appreciated.
Thank you for your time and assistance.
Best regards,
Sotiris