I an facing a problem with -ve enregy of phonon dispersion in MoB2 material.
Here is my input files. File 1 is regular calculation, 4 is denser WFK file generation and 5 is for eph calculation. file 2 and 3 are merging ddb and dvdb. B2Mo_2.abi (511 Bytes) B2Mo_3.abi (2.5 KB)
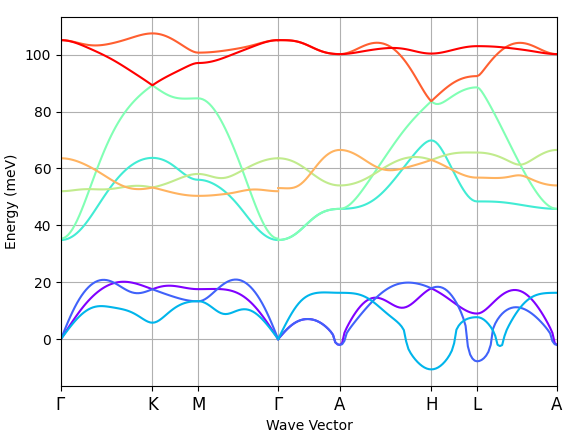
Before I used occopt 7 where I had -ve energy along all the K-path and then I changed it to occopt 3 where I get only -ve energy along A–H–L–A. Please see this attachment below.
Looking on the first output file (B2Mo_1.abo), GS calculation is well converged and also others. Any suggestions to improve this issue would be highly appreciable. Also, I could not quite understand why the DS1 has potential files POT13 and POT15 and why the other DSx files have 1-6 and sometimes1,3,4,6 POT files. Is it because of hexagonal symmetry operations?
it could very well be physical: if your system is unstable, you will get imaginary phonon modes. One way to check is to freeze the corresponding displacements into the structure (you can try freeze_displ in anaddb, or the “agate” utility which is quite powerful) and relax the structure with ionmov 2. If the energy decreases and the relaxation moves away from your initial structure, it was unstable.
Your change of the occopt was an improvement, but it’s a bit strange that it depends so strongly on the specific smearing. Check the smearing width as well - it should be small enough to not perturb things, and occopt 7 is less broad than Fermi Dirac, and should be closer to the true T=0 ground state. You may get artificial instabilities if other things are not converged (ecut, kpt, sometimes evil pseudopotentials, but this has become rare). Check these first to see if the H point phonons move (just 1 DTSET). The stablest values are not necessarily the most physically correct, need to be a bit critical.
If the instability is real, and you know the phase is stable experimentally, you may need to use anharmonic methods to stabilize it, such as (a)TDEP. See the corresponding tutorial
or the TDEP code
As to the numbering of the POT files, DTSET1 does the E field perturbations as well. The others just irreducible atomic perturbations. See the tip
to understand the ipert and idir indices which give the suffix number.
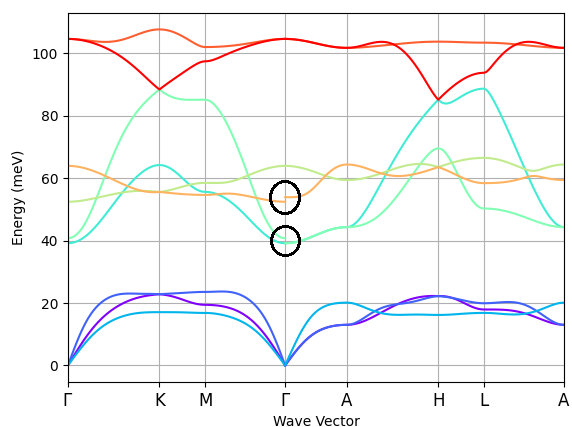
Thanks for the detailed explanation. I checked the structure with relaxation method and It was very close to the stable structure. Then I increased tsmear to 0.03 and looks like it gives reasonable phonon bands (see the attached picture)! However, what is happening at Gamma point (marked circle) I don’t understand. Why is there a gap between those two marked bands? Is it something related to k-points slicing or interpolation of the eph matrix elements? How can I improve this?
Thanks,
Rajesh
This is perfectly physical - you are seeing the non-analytical behavior of the phonon frequencies in presence of LO/TO splitting. Depending on the direction of arrival of the phonon q vector, the macroscopic longitudinal E field will be polarized in different directions and produce a different macroscopic polarization.
Please see the tutorials on DFPT and classical litterature, e.g. Xavier’s prb 55 10355 (1997), and their refs 39 and 27 (Pick Martin and Cohen + the QE team)
I have a one question regarding number of bands. How do I calculate the number of bands need to be considered for the calculation? When I don’t put the nband command in the input file it takes mband=12. Is there any way to select that number? Or do I need to check the GS convergence test with different band number? I guess this number depends on the material characteristic whether it is metal or insulator/semiconductor.