I was trying to calculate the phonon dispersion of monolayer and bulk VTe2. The bulk result looks reasonable, but the monolayer result looks strange and totally different. The only difference between two files is that for the monolayer file I multiply the c lattice constant (z direction) by four times and divide the z component of the position of each atom by four times to get a vacuum layer.
Does anyone know the reason why this cannot work?
Hope someone can help my problem. Thank you!
Here are my steps:
Run abinit VTe2_bulk_1.abi >VTe2_bulk_1.log 2>err & VTe2_bulk_1.abi (4.2 KB)
Run mrgddb < VTe2_bulk_3.abi to combine all DDB files VTe2_bulk_3.abi (749 Bytes)
Run anaddb < VTe2_bulk_5.files > VSe2_1_5.log VTe2_bulk_5.abi (4.1 KB)
4.VTe2_bulk_5.files
VTe2_bulk_5.abi
VSe2_bulk_5.abo
VSe2_bulk_3.ddb.out
VSe2_bulk_5_band2eps
VSe2_bulk_dummy1
VSe2_bulk_dummy2
VSe2_bulk_dummy3
5. Run abiopen.py VTe2_bulk_5_PHBST.nc --expose --seaborn=talk to plot the phonon dispersion bands
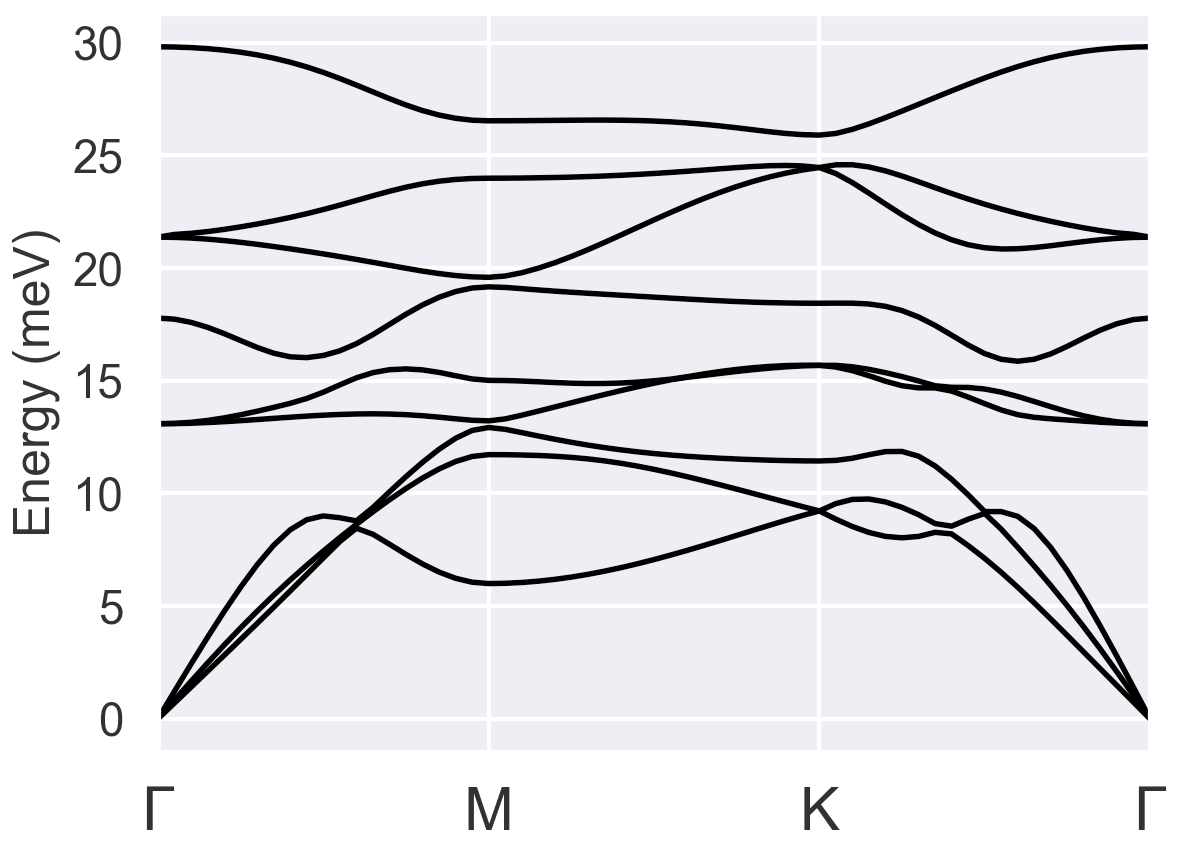
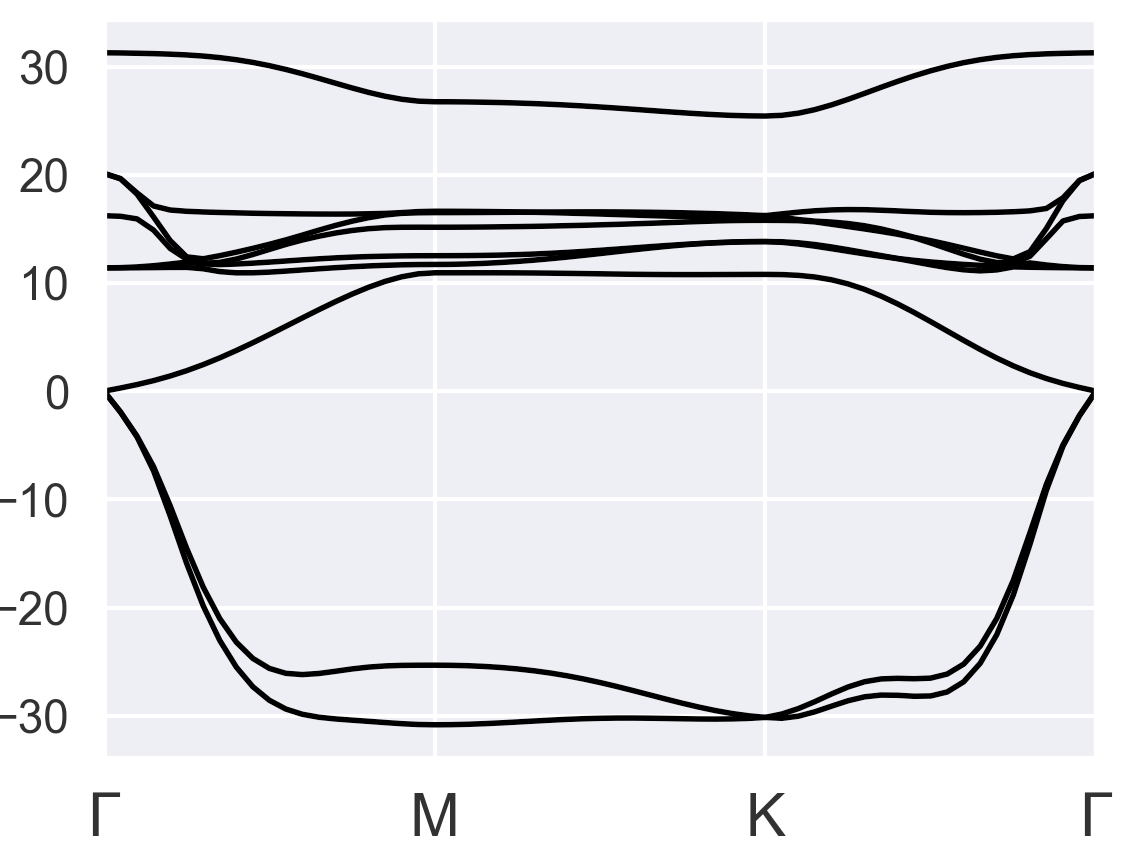
The phonon dispersion of bulk result: (reasonable)
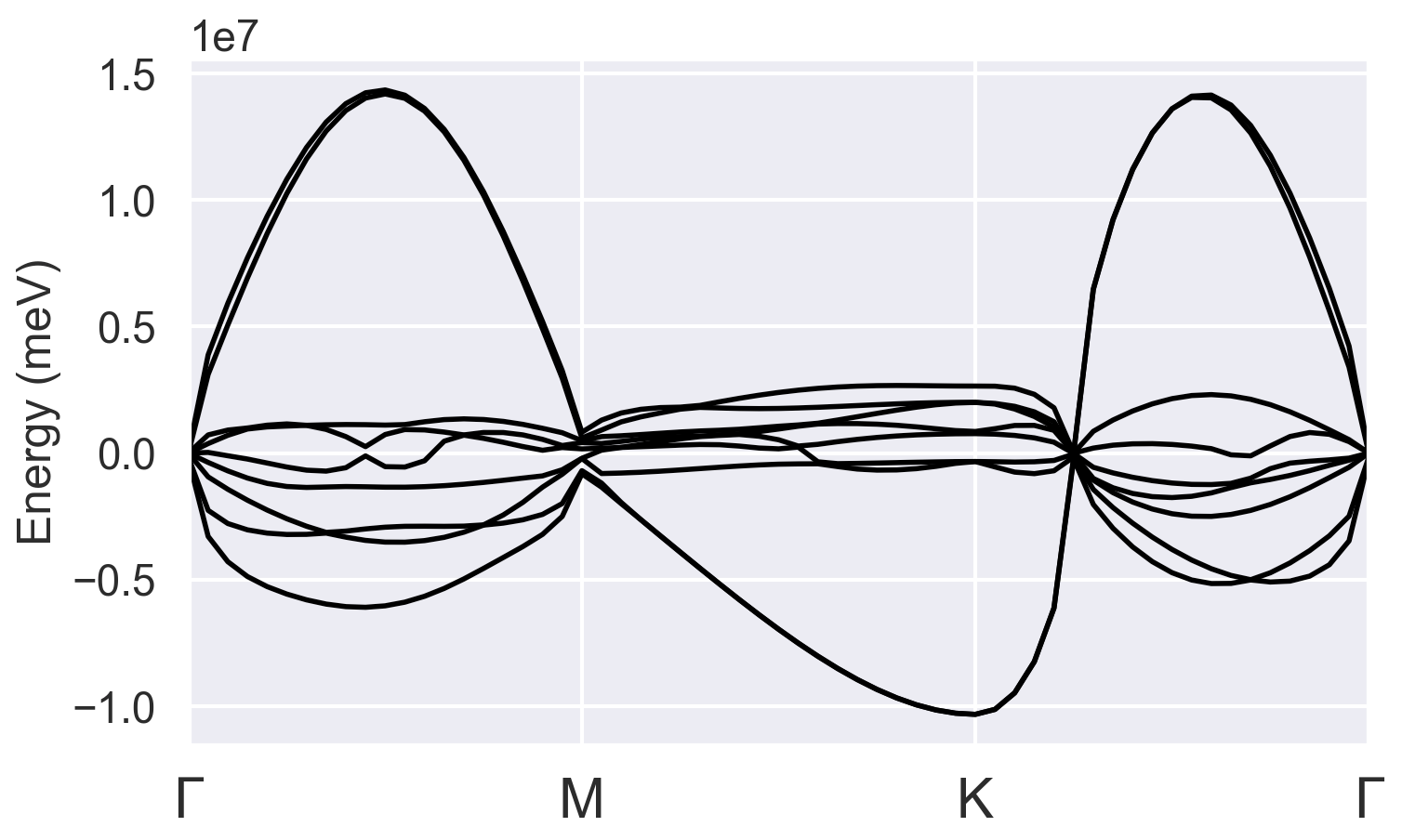
However, after following the same procedure with the files for monolayer VTe2: VTe2_1_1.abi (4.3 KB), the phonon dispersion plot becomes:
I noticed the total energy doesn’t converge and residm keeps increasing in this case. I don’t know what parameters in the input files were not defined well. Thank you for your help!
It seems from your log files that the convergence in the response function calculation goes completely erratic.
One general aspect that should be corrected is to allow for the calculations to reach convergence by increasing the number of steps. In all cases it seems that the calculations never reach the desired convergence target.
Since you are dealing with a metallic phase, personally, I would use a physical electronic temperature (Fermi-Dirac).
If other issues will arise I would suggest this older post PAW+DFPT+GGA has unreasonable energy values - #5 by baguetl
Please let us know if it doesn’t work out.
Bogdan
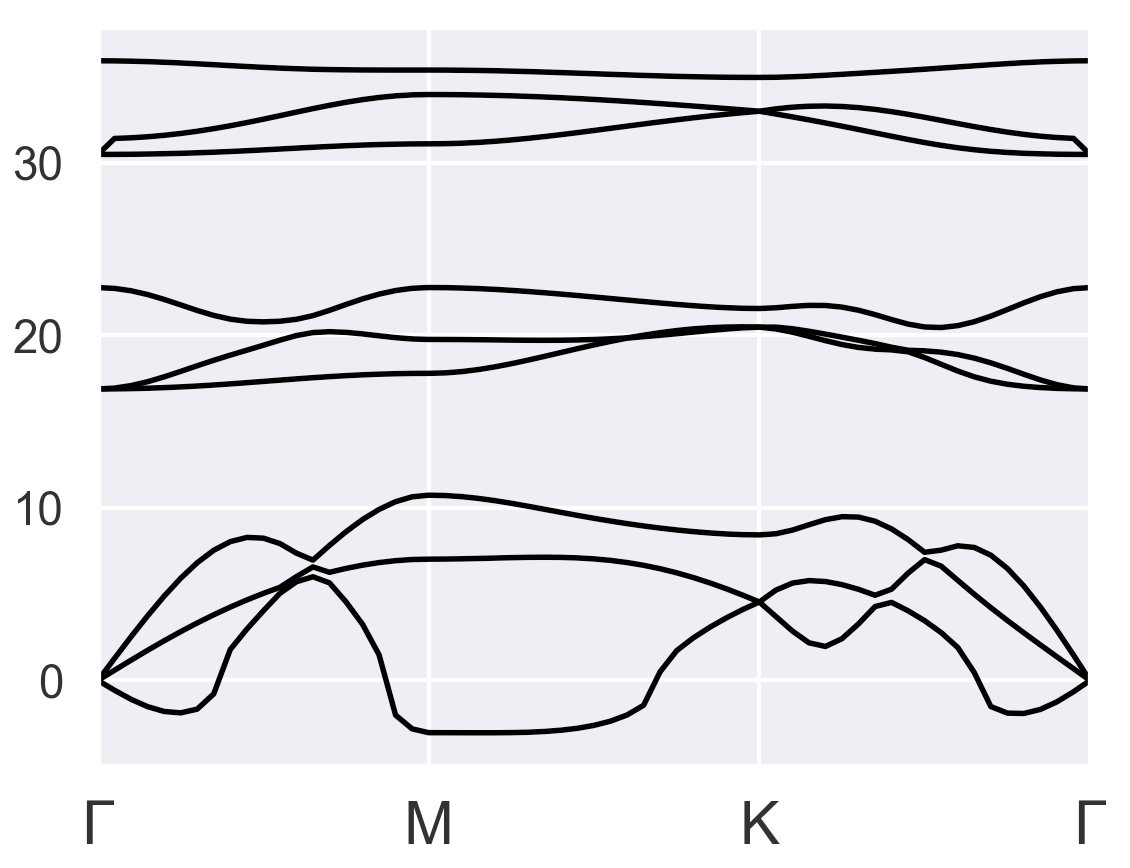
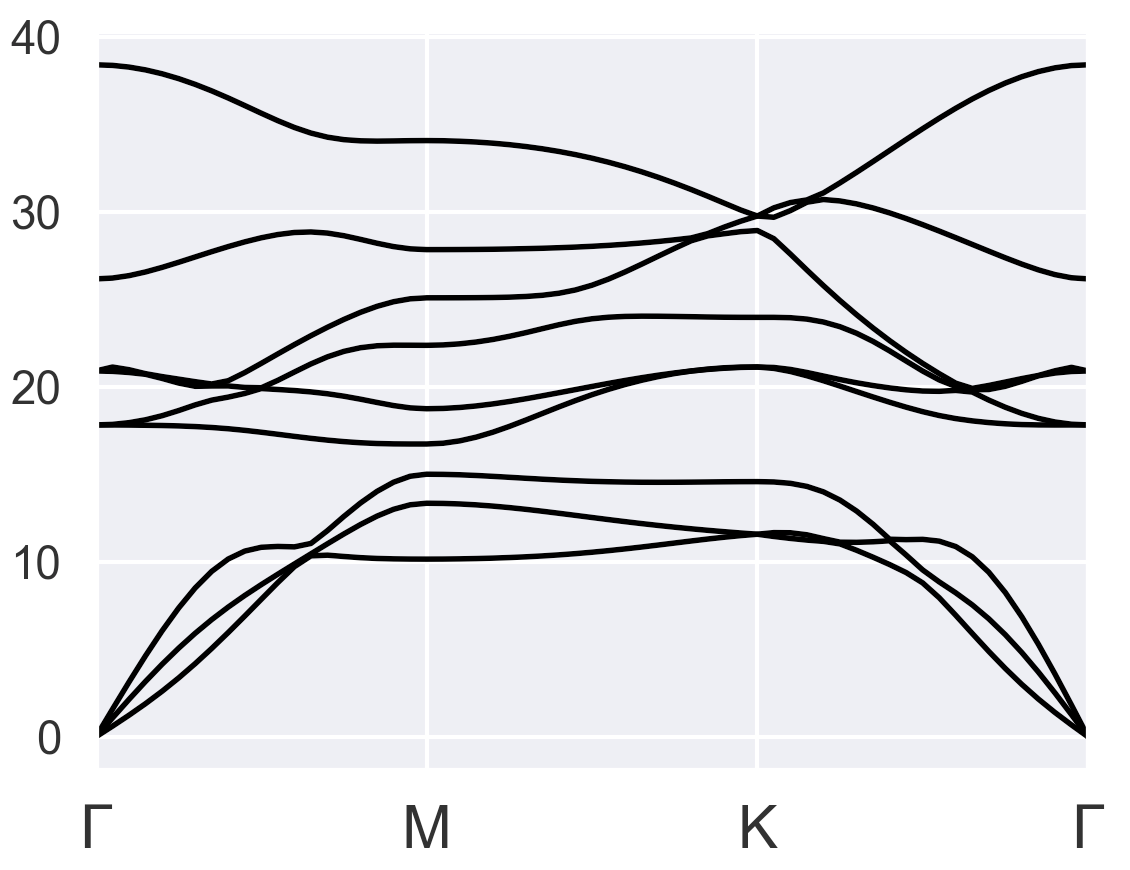
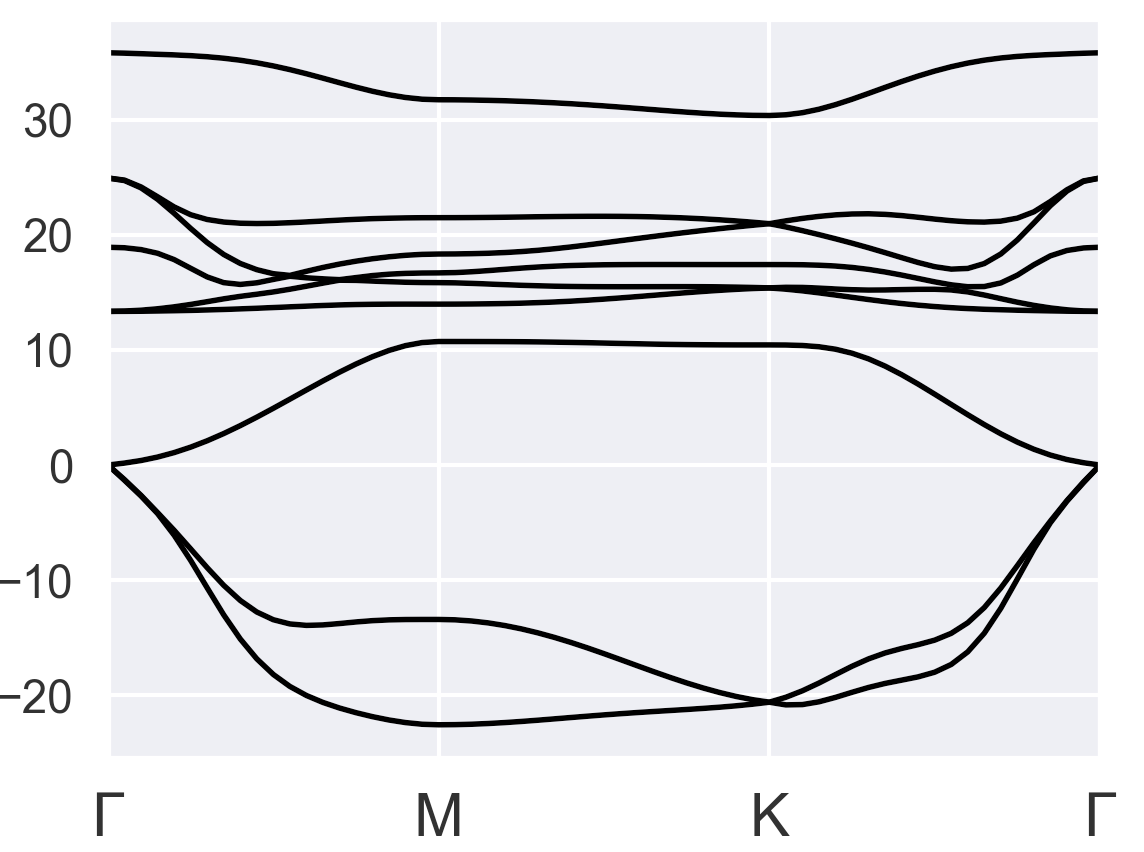
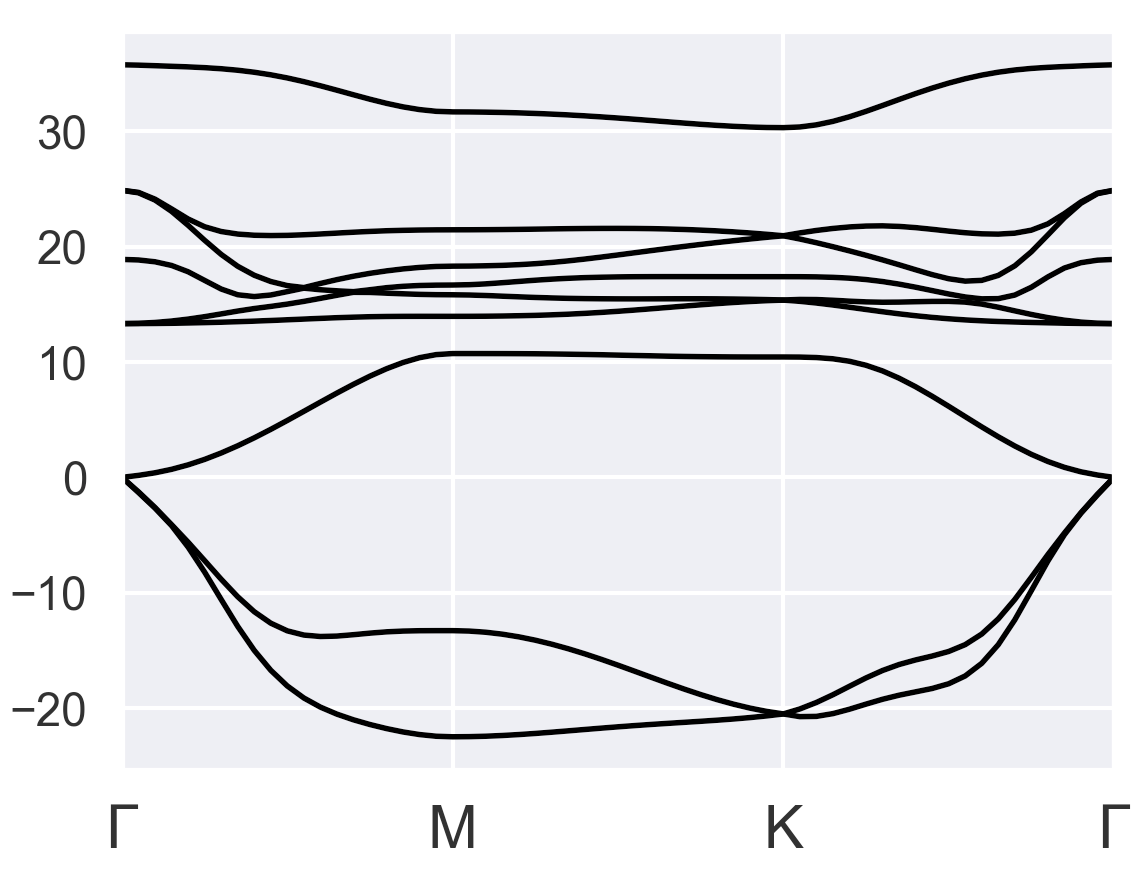
I gave several tries, but none of them looks okay. The first thing I noticed is the total energy started to converge after I changed pseudopotentials from PAW to LDA or GGA as suggested in the older post PAW+DFPT+GGA has unreasonable energy values - #5 by baguetl. I chose occopt = 4 instead of occopt = 3 (Fermi-Dirac for metal) because occopt = 4 seems to be more reasonable than occopt = 3 for the bulk results:
Bulk with occopt = 3 (strange)
xred
V 0 0 0.5
Te 1/3 2/3 0.245
Te 2/3 1/3 0.755
Bulk with occopt = 4 (reasonable)
xred
V 0 0 0.5
Te 1/3 2/3 0.245
Te 2/3 1/3 0.755
Hence, I used occopt = 4 for the following tests: Try1-3 of monolayer VTe2: Pseudopotential = LDA/ occopt = 4
xred (optcell = 0)
V 0 0 0.5/4
Te 1/3 2/3 0.2396/4
Te 2/3 1/3 0.7604/4 VTe2_1_1_try2.abi (4.4 KB) VTe2_1_1_try2.log (2.0 MB)
xred (Te is farther away from V)
V 0 0 0.5/4
Te 1/3 2/3 0.2326/4
Te 2/3 1/3 0.7674/4
xred (Te is closer to V, bulk’s xred)
V 0 0 0.5/4
Te 1/3 2/3 0.245/4
Te 2/3 1/3 0.755/4
Try4 of monolayer VTe2: Pseudopotential = GGA/ occopt = 4
xred (optcell = 0)
V 0 0 0.5/4
Te 1/3 2/3 0.2396/4
Te 2/3 1/3 0.7604/4 VTe2_1_1.abi (4.4 KB) VTe2_1_1.log (2.1 MB)
All results show negative frequencies for a broad range of k-space. Do you have other suggestions for getting a better result?
Thank you!
Looking at the two treatments with the two different types of occopt might point out the fact the occopt 4 is converging much faster in the self consistent cycles (reason for which was intended as far as I know when compared to the Fermi-Dirac smearing), so I am not completely sure if the Fermi-Dirac smearing is wrong, but rather unconverged.
Coming to your other figures regarding the single-layers, I noticed quite a large amount of WARNING messages inside your log files and mostly come from the fact that the convergence with respect to the perturbations is not met (in some cases I have noticed that they are quite far off from your tolerance with more than 7 orders of magnitude higher). For this, a simple fix is to increase the number of steps to allow for the cycle to meet your tolerance (see nstep variable).
Also, a good check for future reference is to inspect the forces on the atoms in the preliminary wavefunction calculations (in your case DATASET 1) before reaching the perturbative part (check your output file for this). In your log files you have information about the Stress (or internal pressure if you prefer) which looks relatively high. Normally this points out that the lattice parameter minimization is missing which might add up to the erroneous phonon dispersion.
An important aspect that you might want to consider is that for DFPT is crucial to have very well converged wavefunctions, and in the presented examples I am not sure if this is the case (like converged ecut, kpoints, very low SCF tolerance…)
I hope this helps.
Good luck and let us know if this helped or not!
Bogdan
How can I optimize lattice parameters (a and b) and atomic positions of a monolayer using optcell and ionmov variables? Is there a different way to optimize monolayers compared to bulk?
Replace PAW pseudopotential with NC. I observed a similar problem for MoS2 but also for graphene. Using PAW for 2D materials (such with artificial vacuum) it is very difficult to get convergent second derivatives.
Hi, many thanks.
I used psudopotentioal in work. But, I do not know how can I have an input file for monolayer optimization. Is it similar to bulk ( for example: optcell 2, ionmov 2)? Else, what must I do?
Yaser
I optimized monolayer materials with the similar method for bulk optimization (the attached file). I am in the learning stage, so I am not sure if it is correct. VTe2_cell.abi (1.2 KB)
Your approach would be ok to use optcell 2 and ionmov 2 for a full relaxation of the crystal structure implying both atomic positions and lattice vectors even in the single-layer, although if you did a preliminary convergence with respect to vacuum, one might use one of the other optcell options (7,8 or 9) where one can fix the vacuum distance while relaxing only the in-plane lattice vectors.
Just to clarify further, there is no harm in using optcell and ionmov 2, and if you do so, you might find a slight deviation from your initial vacuum distance estimation if the convergence of vacuum was done properly, so in a way, you can use it as a double-check that your vacuum assessment was correct. Computationally, by not fixing the vacuum distance with optcell 7,8 or 9, you add an extra degree of freedom to be considered by the forces minimization algorithm, so if you deal with few degrees of freedom, for example, fixed atoms positions (as validated by their Wyckoff positions) and equivalent in-plane lattice vectors, instead of one degree of freedom, you will end up with two degrees of freedom, thus finally slightly increasing the number of steps to reach the desired relaxed structure.
I hope this helps, please let me know otherwise.
Cheers,
Bogdan
PS For the future, please open a new thread if you don’t find a solution among the already discussed issues that other users might face.