Dear all:
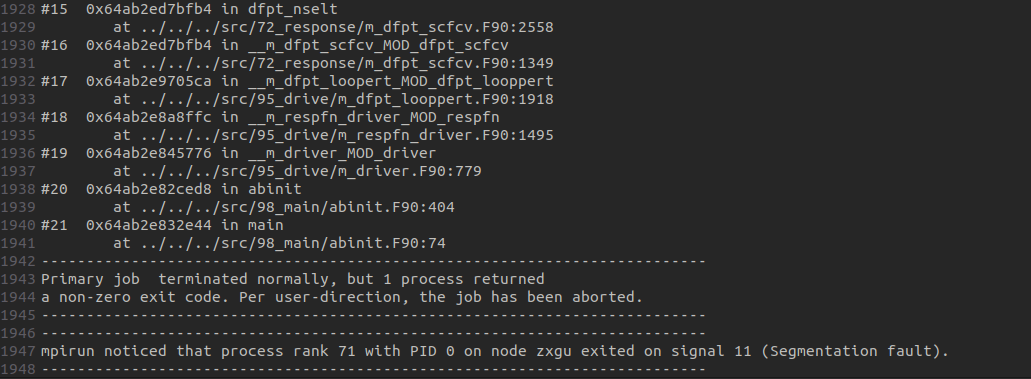
I am trying to do a piezoelectric tensor calculation following the tutorial elastic - abinit using Abinit 10.4.5.. Now in step 2 (calculation of several 2nd order derivatives of the total energy) it seems Ok until the perturbation reaches the last atom and the calculation stopped without outputting any DDB file? Please see the pasted .abi and .log files. Similar situation occured twice after a one-week run of the program. Thank you.
#KGN step 2
ndtset 3
###############################################################################
Set 1 : Initial self-consistent run
###############################################################################
kptopt1 1
tolvrs1 1.0d-18 #need excellent convergence of GS quantities for RF runs
########################################################################
Set 2 : Calculate the ddk wf’s - needed for piezoelectric tensor and Born effective charges in dataset 3
#######################################################################
getwfk2 -1
iscf2 -3 #this option is needed for ddk
kptopt2 2 #use time-reversal symmetry only for k points
nqpt2 1 #one wave vector will be specified
qpt2 0 0 0 #need to specify gamma point
rfelfd2 2 #set for ddk wf’s only
tolwfr2 1.0d-20 #only wf convergence can be monitored here
#############################################################################
Set 3 : response-function calculations for all needed perturbations
############################################################################
getwfk3 -2
getddk3 -1
kptopt3 2 #use time-reversal symmetry only for k points
nqpt3 1
qpt3 0 0 0
rfphon3 1 #do atomic displacement perturbation
rfstrs3 3 #do strain perturbation
tolvrs3 1.0d-10 #need reasonable convergence of 1st-order quantities
##################################################################
Common input data
##################################################################
acell 1.9630015360E+01 1.8731783186E+01 3.0242540769E+01 Bohr
rprim 9.9998997866E-01 0.0000000000E+00 4.4768932538E-03
0.0000000000E+00 1.0000000000E+00 0.0000000000E+00
-1.7400338913E-01 0.0000000000E+00 9.8474505359E-01
#Definition of the atom types and atoms
ntypat 4
znucl 8.00000 7.00000 6.00000 1.00000
natom 156
typat 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 3 3 3 3
3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3
3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3
3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3
3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 4 4 4 4
4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4
4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4
4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4
xred ####################COPY RELAXED RESULT FROM PREVIOUS CALCULATION
2.0242339844E-01 3.0532617535E-01 5.0137591005E-01
7.9757660156E-01 8.0532617535E-01 4.9862408995E-01
……..
#Gives the number of bands, explicitely (do not take the default)
nband 248 # For an insulator (if described correctly as an
insulator by DFT), conduction bands should not
be included in response-function calculations
#Definition of the plane wave basis set
ecut 22.0 # Maximum kinetic energy cutoff (Hartree)
ecutsm 0.5 # Smoothing energy needed for lattice paramete
optimization. This will be retained for
consistency throughout.
#Definition of the k-point grid
kptopt 1 # Use symmetry and treat only inequivalent points
ngkpt 1 1 1 # 1x1x1 Monkhorst-Pack grid (gamma)
nshiftk 1 # Use one copy of grid only (default)
shiftk 0.0 0.0 0.0 # This choice of origin for the k point grid
preserves the hexagonal symmetry of the grid,
which would be broken by the default choice.
#Definition of the self-consistency procedure
diemac 3.0 # Model dielectric preconditioner (model DIElectric MACroscopic constant)
#, For wider gap insulators, use 2.0 … 4.0
nstep 600 # Maxiumum number of SCF iterations
enforce calculation of forces at each SCF step
optforces 1
pp_dirpath “$ABI_PSPDIR”
pseudos “O.psp8, N.psp8, C.psp8, H.psp8”
KGN_elast2_1_248band.log (2.8 MB)