Processing: mos2.out…
Processing: mos2.in…
Dear abinit users,
I try to calculate gamma point phonon in bilayer MoS2 but the potential residual diverges. Any help is appreciated.
Regards,
Koyendrila
Hello,
you need to give quite a bit more details for us to be able to help. input, machine architecture etc…
If you are doing a bilayer in vacuum, I have noticed that the DFPT sometimes has trouble with GGA. Try LDA to see if that is the issue. For some reason the low density in the vacuum makes things diverge.
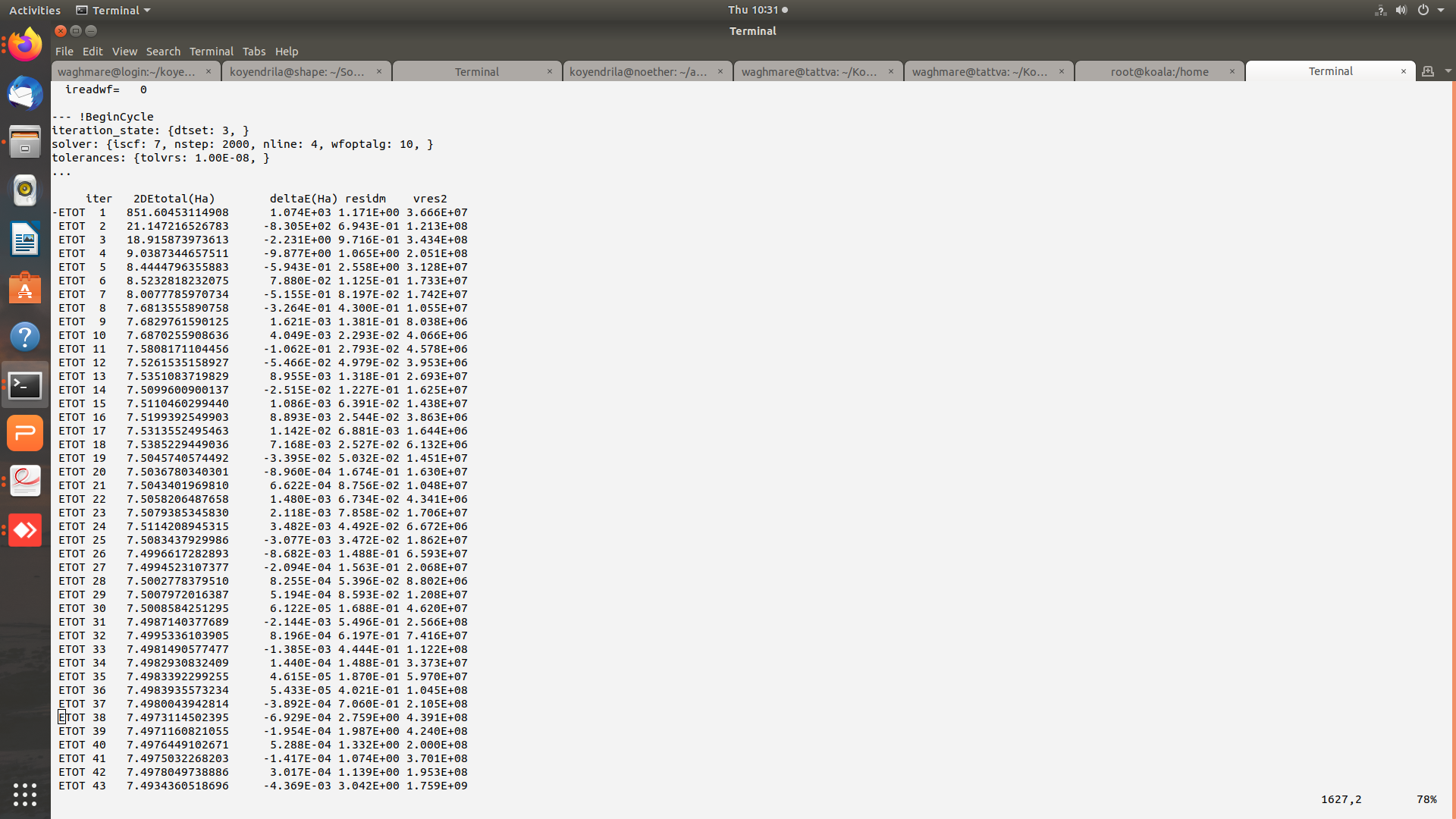
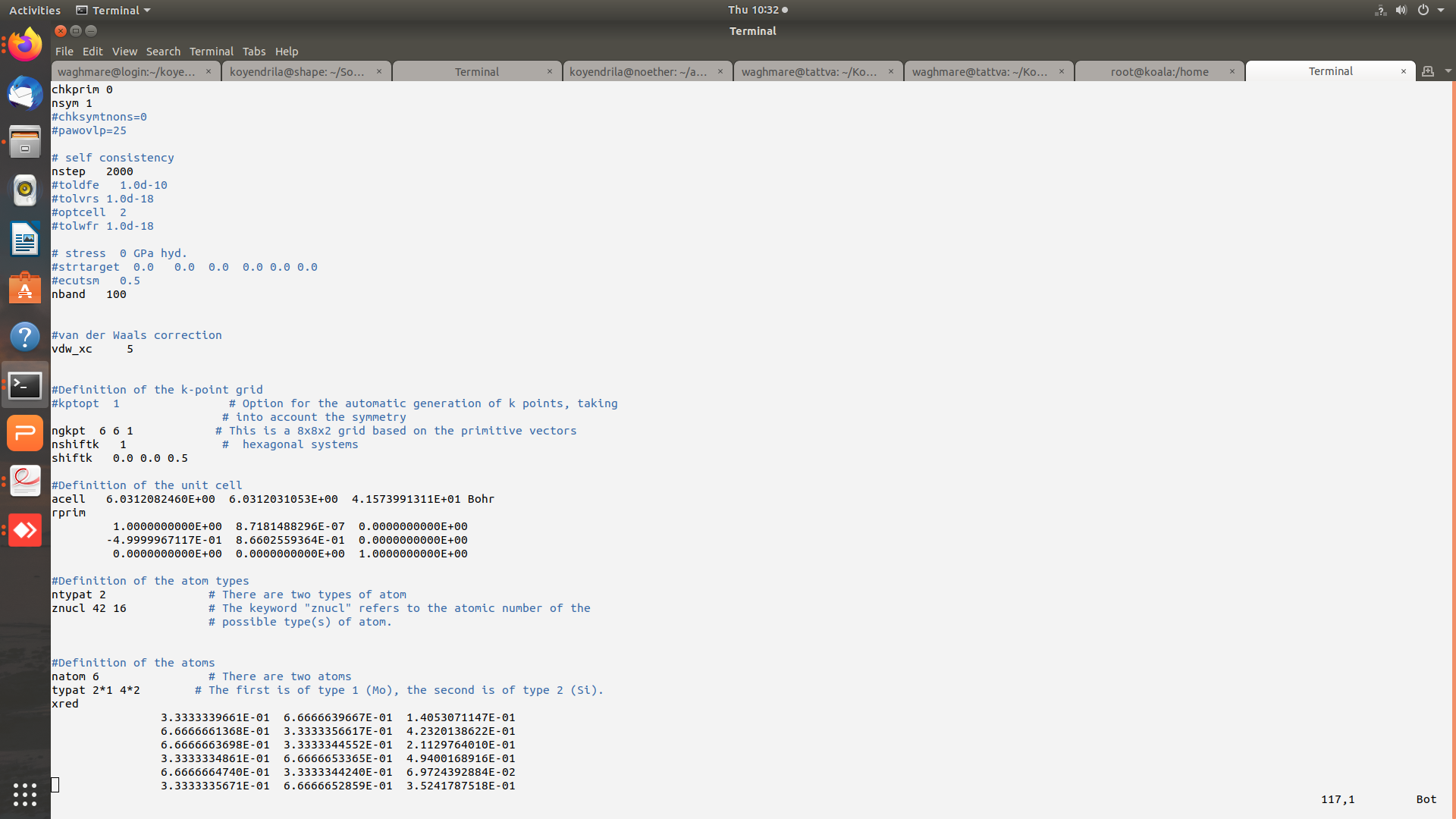
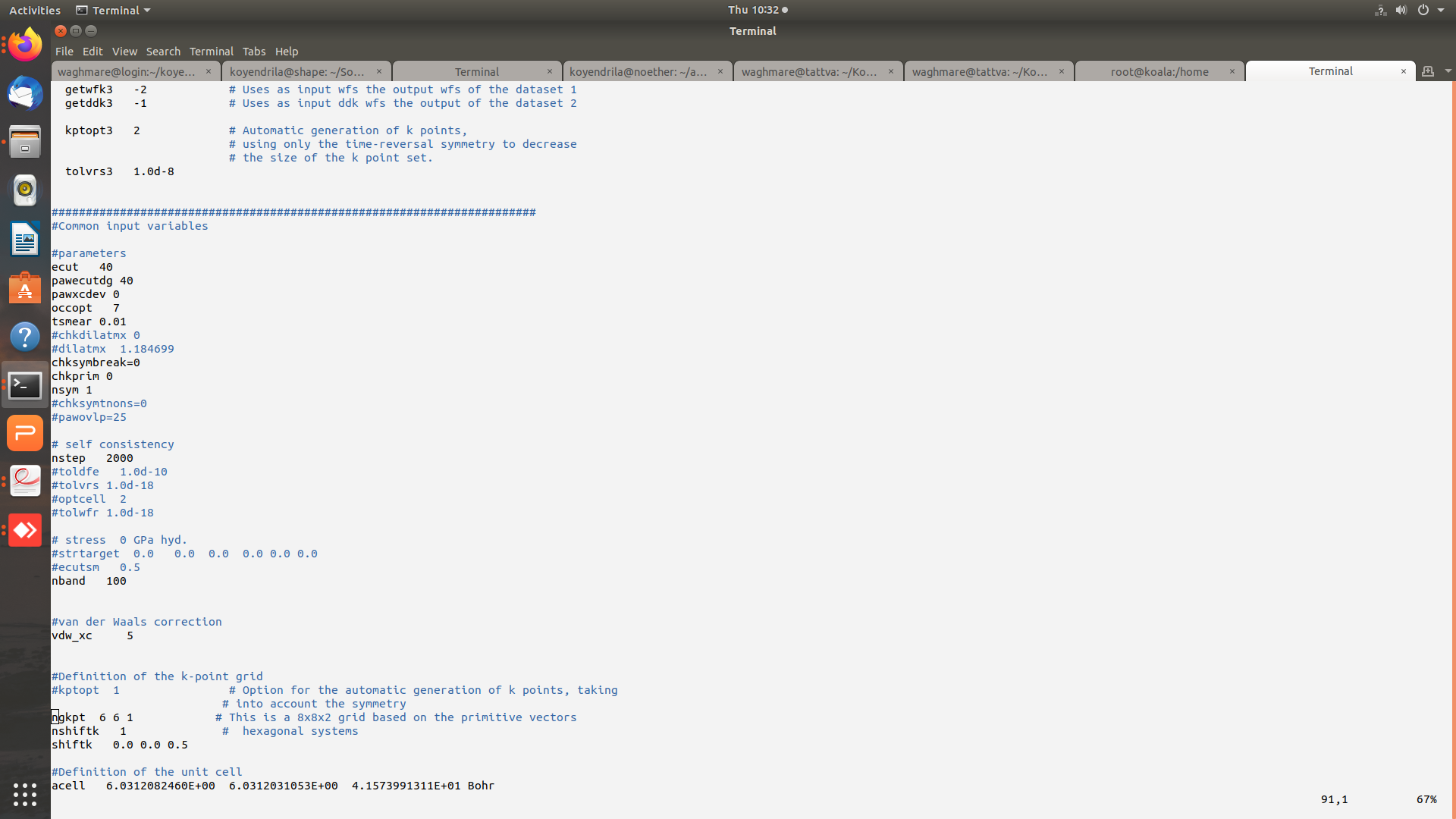
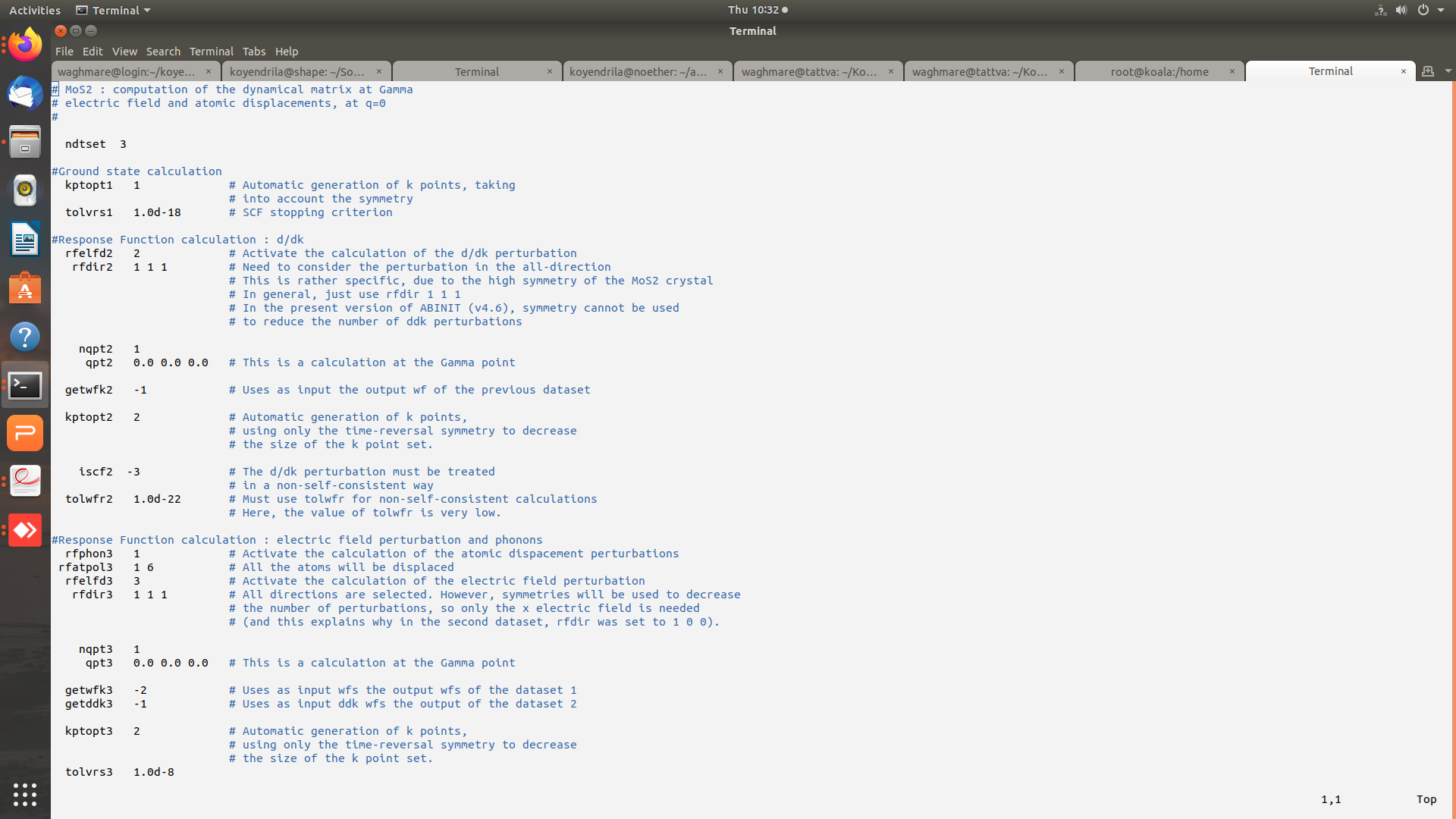
Hi Mverstra,
Thanks for replying. Plesae find attached the images of input (and output) files for the gamma point phonon calculation. The potential residual diverges (tolvrs ~ 3.486E+58 at the end of 464 nsteps). I am presently working with GGA-JTH pseudopotentials and have even tried Norm-conserving PBE PSP8 pseudo but the problem persists.
The forum doesn’t allow me to upload the files (taking a look at the files would be helpful).
Regards,
Koyendrila
JNCASR
Dear Matthieu,
I have followed this link (No convergence in ddk - ABINIT Discussion Forums) and used occopt 7 and tsmear = 0.002 and included few extra bands bit it’s not working.
A part of the input file is attached:
#Common input variables
#parameters
ecut 40
pawecutdg 40
pawxcdev 0
occopt 7
tsmear 0.01
#chkdilatmx 0
#dilatmx 1.184699
chksymbreak=0
chkprim 0
nsym 1
#chksymtnons=0
#pawovlp=25
self consistency
nstep 2000
#toldfe 1.0d-10
#tolvrs 1.0d-18
#optcell 2
#tolwfr 1.0d-18
stress 0 GPa hyd.
#strtarget 0.0 0.0 0.0 0.0 0.0 0.0
#ecutsm 0.5
nband 100
#van der Waals correction
vdw_xc 5
#Definition of the k-point grid
#kptopt 1 # Option for the automatic generation of k points, taking
# into account the symmetry
ngkpt 6 6 1 # This is a 8x8x2 grid based on the primitive vectors
nshiftk 1 # hexagonal systems
shiftk 0.0 0.0 0.5
#Definition of the unit cell
acell 6.0312082460E+00 6.0312031053E+00 4.1573991311E+01 Bohr
rprim
1.0000000000E+00 8.7181488296E-07 0.0000000000E+00
-4.9999967117E-01 8.6602559364E-01 0.0000000000E+00
0.0000000000E+00 0.0000000000E+00 1.0000000000E+00
#Definition of the atom types
ntypat 2 # There are two types of atom
znucl 42 16 # The keyword “znucl” refers to the atomic number of the
# possible type(s) of atom.
#Definition of the atoms
natom 6 # There are two atoms
typat 21 42 # The first is of type 1 (Mo), the second is of type 2 (Si).
xred
3.3333339661E-01 6.6666639667E-01 1.4053071147E-01
6.6666661368E-01 3.3333356617E-01 4.2320138622E-01
6.6666663698E-01 3.3333344552E-01 2.1129764010E-01
3.3333334861E-01 6.6666653365E-01 4.9400168916E-01
6.6666664740E-01 3.3333344240E-01 6.9724392884E-02
3.3333335671E-01 6.6666652859E-01 3.5241787518E-01
The convergence problem in potential residue is persistent.
Regards,
Koyendrila
Hi again,
try LDA - I had something similar with bilayer MoS2. Need to debug the DFPT vxc part for GGA with vacuum, but this is longer term.
Other stuff:
- You should never need so many scf steps. Beyond 100 something is wrong.
- pawecutdg is a bit low compared to ecut (ratio should be at least a factor of 1.2 or something like that). ecut seems a bit high for PAW potentials.
- why do you remove symmetries? The atomic positions are a bit crappy - are they relaxed properly? Otherwise the phonons will not be very representative
- the markdown does not render typat properly
- you can set diemac to something low, 1-4 or so to precondition the SCF cycle, just like in GS calculations
- I would avoid the non zero shiftk, especially if symmetry is disabled: in a slab shifting along z should have no impact
- tsmear 0.01 is a bit high. Should not be a big deal, but might change things
Thanks. The structure relaxes properly in 14 iterations. I could incorporate these suggestions and try running the calculations again. Will it be possible to share the input file and the pseudopotentials for your calculations on bilayer MoS2?
Regards,
Koyendrila
My other file is not useful, it was for testing purposes and severely underconverged for other things (equivalent to yours for the rest apart from vdw_xc which I set to 6, and shiftk which is 0).
GaN_.out.in (178.4 KB)
GaN.OPT.cif.in (1.1 KB)
GaN_TEST.IN (4.6 KB)
Hi Dear ABINIT users,
I have used the following input file (GaN_TEST.IN) as a test to learn how I can calculate phonon frequencies and find the stability of monolayers. As it can be seen from the obtained results (GaN_.out.in), the results do not show the stability of GaN monolayer because of the negative phonon frequencies. These results indicate that the input file has problems. What changes do I need to make to achieve the desired result?
I look forward to receiving your generous assistance.
The best wishes