Hello everyone,
I am a beginner in using ABINIT and recently, I have been learning how to calculate the second-order nonlinear optical properties of materials with it. However, I have encountered some issues during this process, and after several unsuccessful attempts to resolve them on my own, I am hoping someone can assist me.
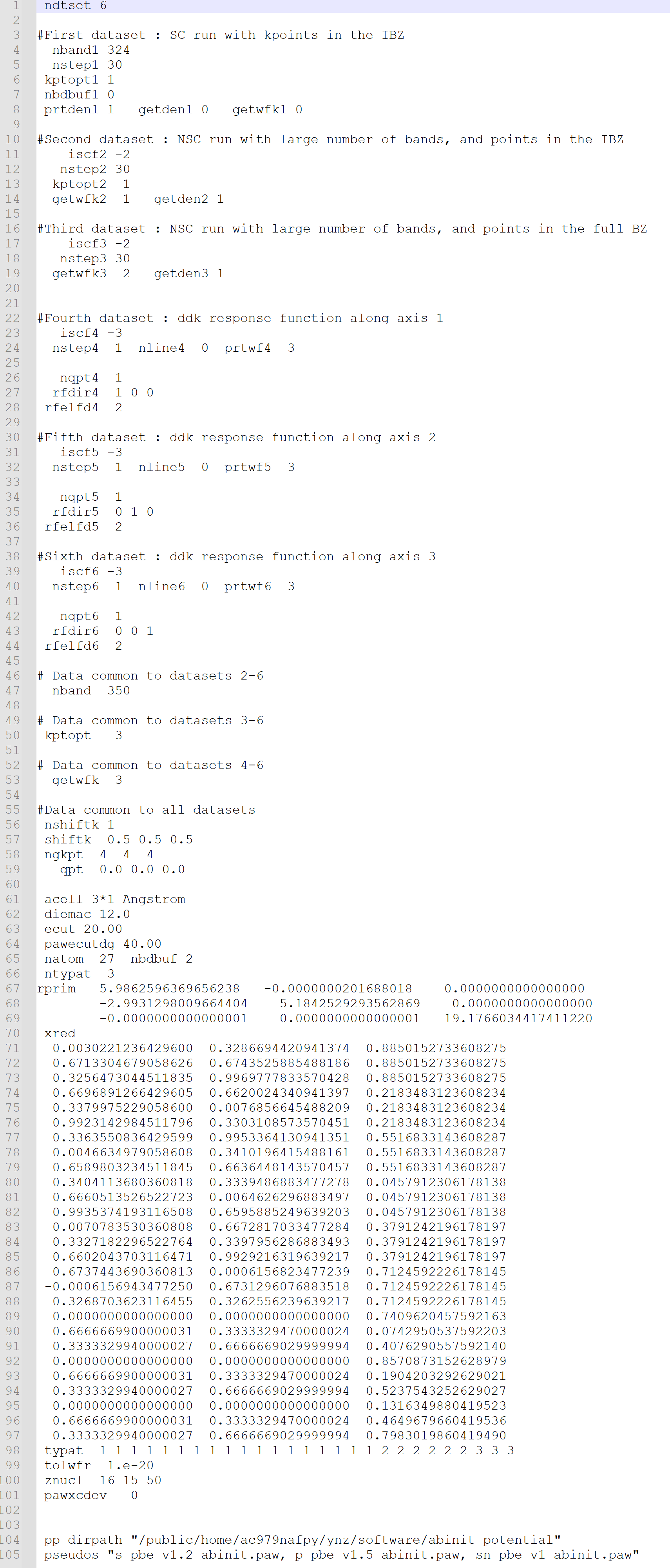
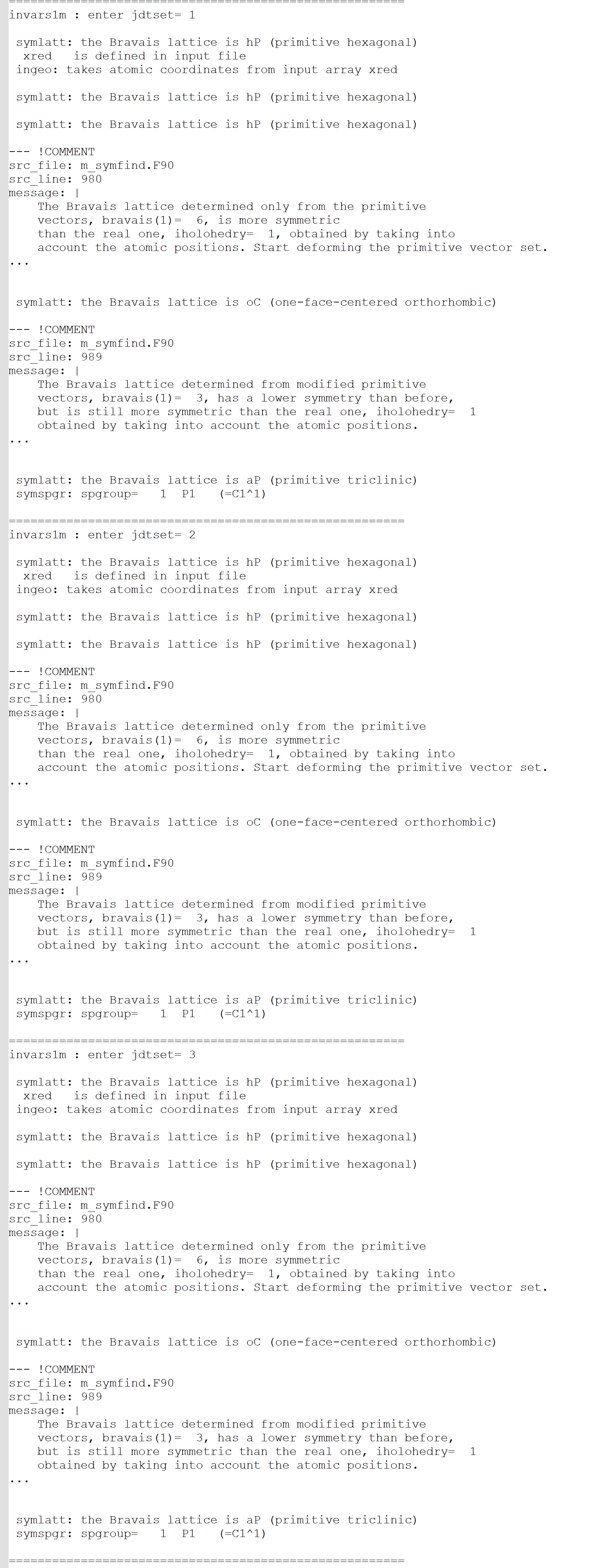
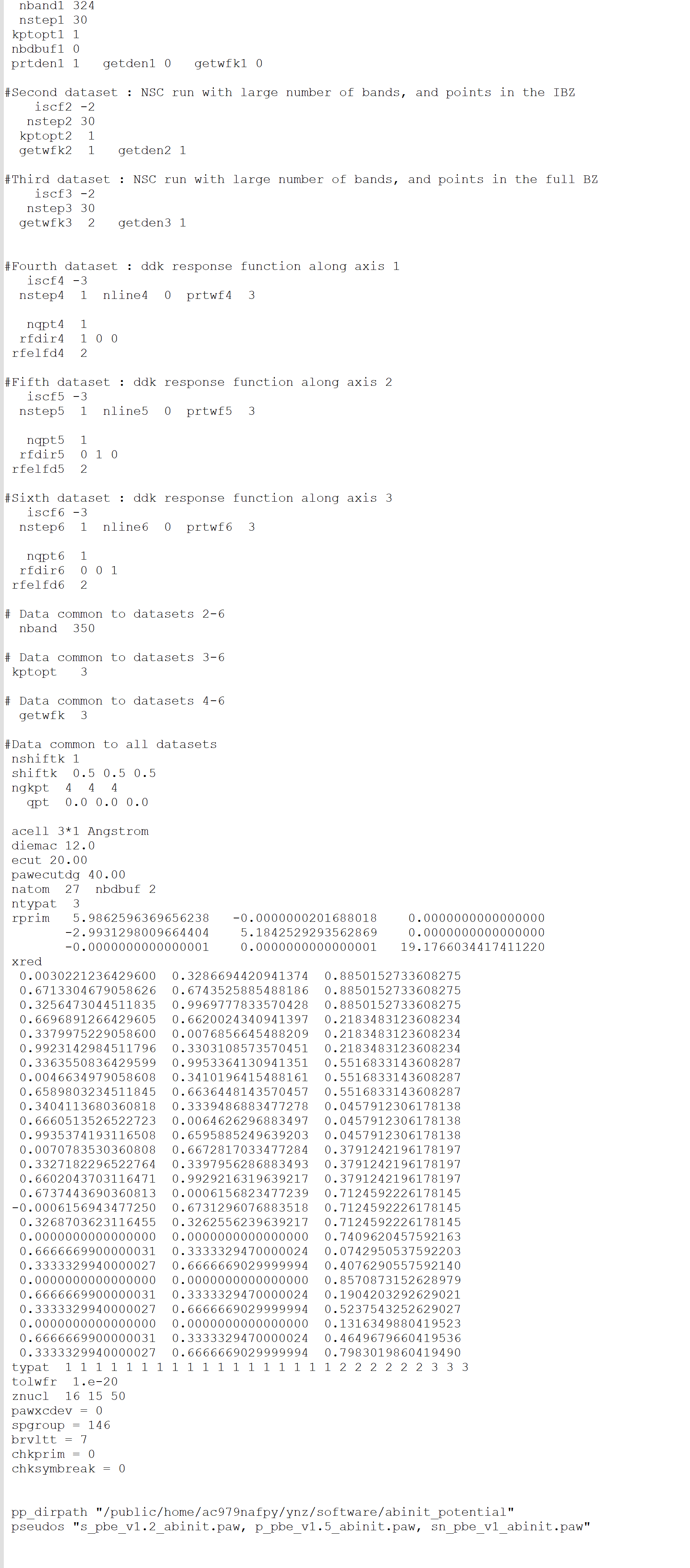
I did not encounter any problems when running the examples provided on the official website, but I faced symmetry issues while testing SnP2S6. The structural parameters of SnP2S6 were obtained using VASP optimization. Upon reviewing the run files, I found that ABINIT was unable to correctly recognize the space group (P146), and instead, it used the space group P1.
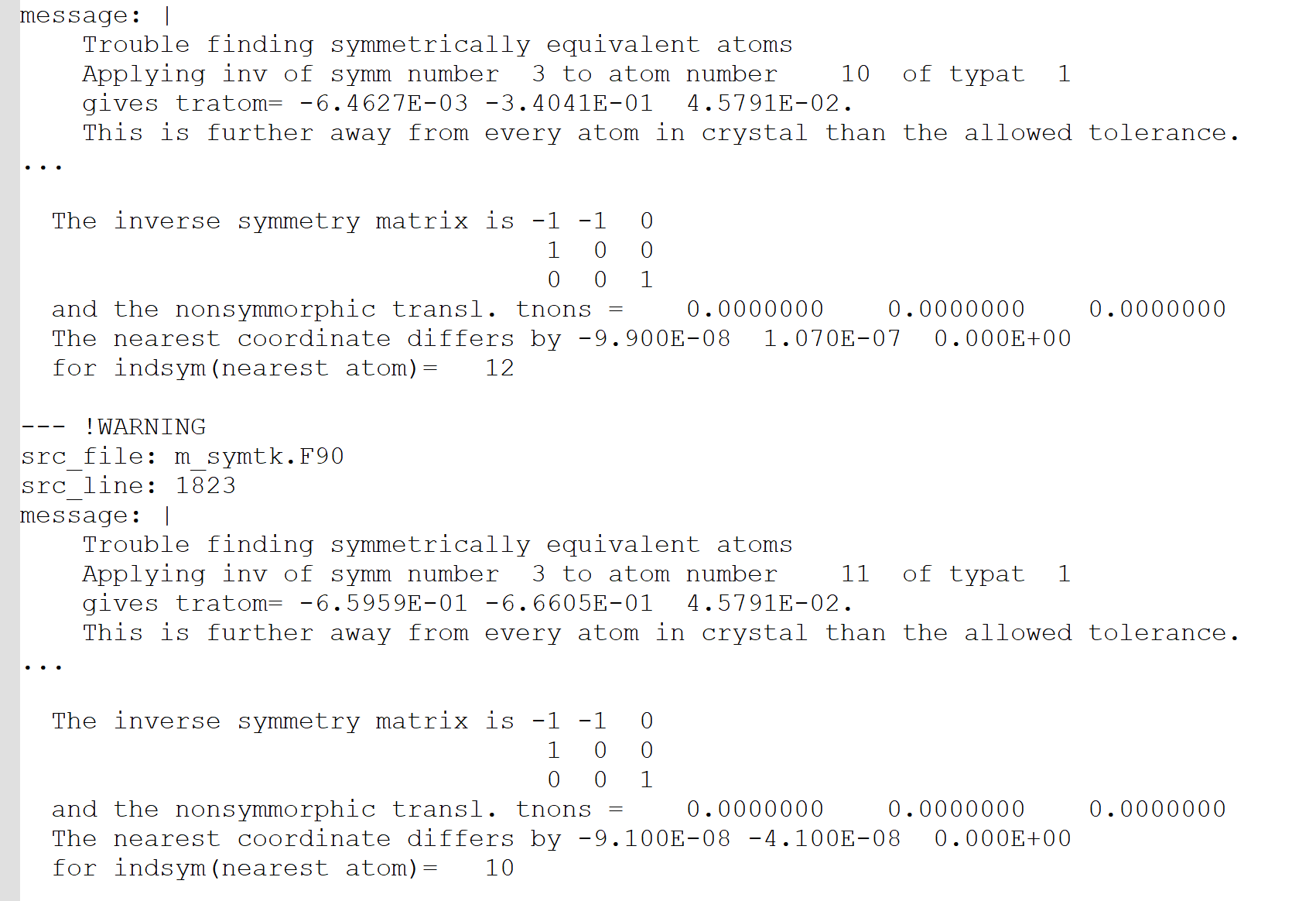
When I specified the space group as 146 using the Spgroup parameter and reran the calculation, ABINIT issued warnings about large errors when identifying equivalent atoms through symmetry. Initially, I did not pay much attention to this. However, after computing the entire second-order susceptibility tensor, I noticed that in theory, certain elements (such as in the ZXY direction) should not be zero, but ABINIT calculated them to be nearly zero.
I earnestly hope that someone with more experience can help me resolve this issue. Any assistance would be greatly appreciated!
I’m sorry, the system prompted that newly registered users are unable to upload files, so I provided my files in the form of pictures.