Hello,
I tried to calculate phonon band structure for fcc lattice. Independently on which parameters I use (k- and q-point grid, with/without shiftk, q-path, material), I get the same wrong behavior around K/U high-symmetry point.
For example, band structure in copper:
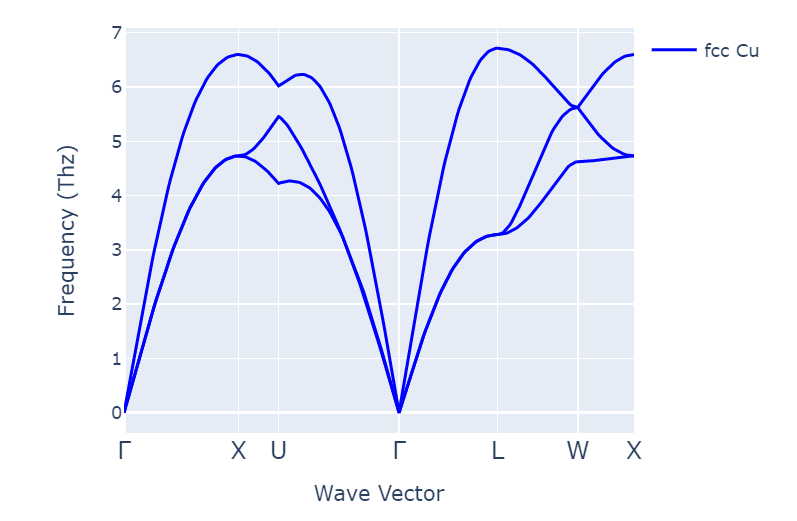
I got the same dispersion following different ways. For example, via phonon workflow:
import os
import sys
import abipy.abilab as abilab
from abipy import flowtkdef make_scf_input(structure, ngkpt, tsmear, pseudos, paral_kgb=1):
scf_inp = abilab.AbinitInput(structure, pseudos=pseudos) scf_inp.set_vars( istwfk="*1", ecut=50.0, nband=16, occopt=3, tsmear=tsmear, nstep=500, paral_kgb=paral_kgb, autoparal=4, wfoptalg=14) # Dataset 1 (GS run) scf_inp.set_kmesh(ngkpt=ngkpt, shiftk=structure.calc_shiftk()) scf_inp.set_vars(tolvrs=1e-10) return scf_inpdef build_flow(options):
if not options.workdir:
options.workdir = os.path.basename(sys.argv[0]).replace(“.py”, “”).replace(“run_”, “flow_”)structure = abilab.Structure.from_abivars( acell=[6.8313599418]*3, rprim=[ [0.0, 0.5, 0.5], [0.5, 0.0, 0.5], [0.5, 0.5, 0.0] ], typat=1, xred=[0, 0, 0], ntypat=1, znucl=29) pseudos = pseudos = "/gpfs/home3/fedorak/pseudos/Cu.psp8" flow = flowtk.Flow(workdir=options.workdir) scf_work = flowtk.Work() ngkpt_list = [[16]*3] tsmear_list = [1e-3] for ngkpt in ngkpt_list: for tsmear in tsmear_list: scf_input = make_scf_input(structure, ngkpt, tsmear, pseudos) scf_work.register_scf_task(scf_input) flow.register_work(scf_work) for scf_task in scf_work: ph_work = flowtk.PhononWork.from_scf_task(scf_task, qpoints=[4]*3, is_ngqpt=True) flow.register_work(ph_work) return flow.allocate(use_smartio=True)@flowtk.flow_main
def main(options):
return build_flow(options)if name == “main”:
sys.exit(main())
With the q-path provided in anaddb file:
ifcflag 1 ! Interatomic force constant flag
brav 2 ! Bravais Lattice : 1-S.C., 2-F.C., 3-B.C., 4-Hex.)
ngqpt 4 4 4 ! Monkhorst-Pack indices
nqshft 1 ! number of q-points in repeated basic q-cell
q1shft 3*0.0
nqpath 7
qpath 0.0000000 0.0000000 0.0000000 !\Gamma
0.5000000 0.0000000 0.5000000 !X
0.6250000 0.2500000 0.6250000 !U
0.0000000 0.0000000 0.0000000 !\Gamma
0.5000000 0.5000000 0.5000000 !L
0.5000000 0.2500000 0.7500000 !W
0.5000000 0.0000000 0.5000000 !X
ddb_filepath = “/abspath_to_workdir/outdata/out_DDB”
output_file = “anaddb.abo”
Hope someone can provide insights what I am doing wrong.