Dear colleagues,
I was trying to generates bands using wannier90 for PrNiO2 system. I am using abinit-9.8.3 and wannier90-3.1.0. DFT band structure works very well. However, bands from wannier90 does not looks good. I tried with different frozen energy window, mp_grid, num_bands but, it does not give good results.
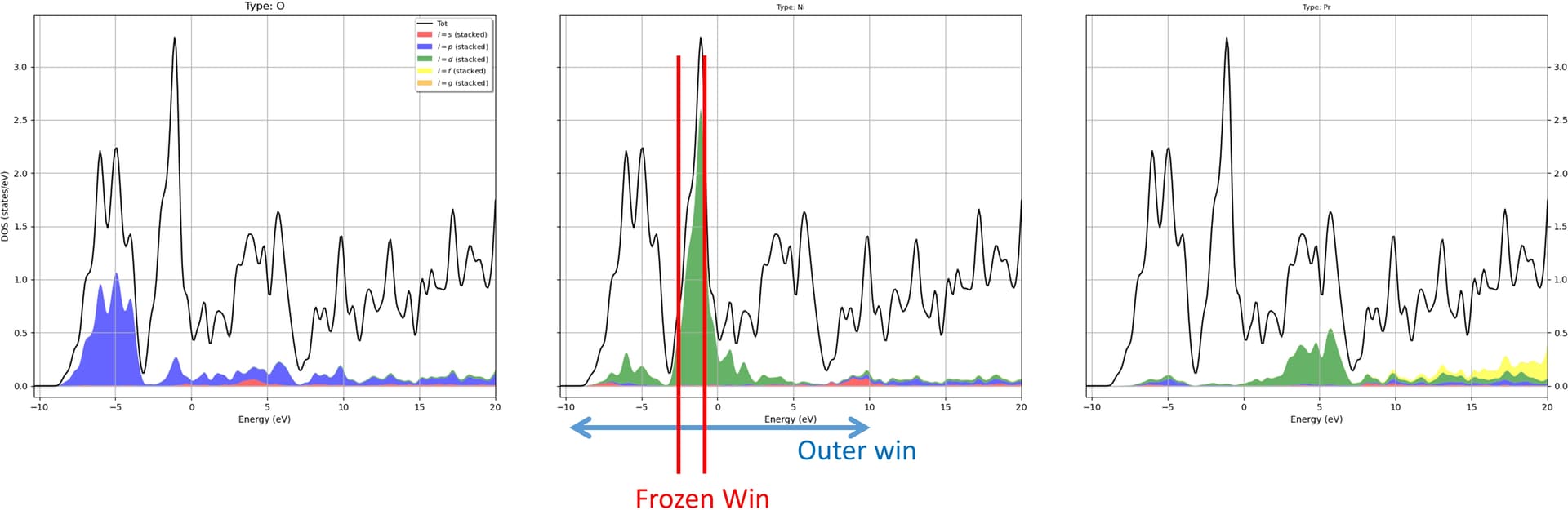
Please have a look at the DOS picture where I am calculating projections for Ni 3d bands and accordingly I chose the frozen window. In the frozen window (FW) there is still O-2p states but I can not increase the FW as it gives error: more states in FW that target WFs!
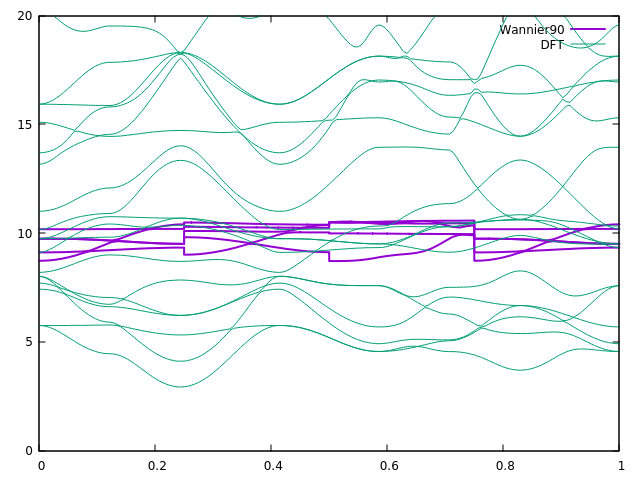
I have attach the band plot here.
Also, here is the input files for wannier90 and abinit. I have followed the tutorial for La2CuO4 system.
PNO.abi (4.1 KB)
PNOo_DS3_w90.win.abi (594 Bytes)
Another point I have noticed that the conv_tol does not reached even by 10000 num_iter, it goes upto 10-7 or -8.
Could someone please have a look and help me how to improve the results? Any suggestions for the wannier90 calculations. Please let me know if you need further info.
Thanks!
Rajesh
There was mistake in the .win file. The begin_kpoint_path should be given left to right on the same line.
Dear All,
I face another problem. Could some one please help me to figure out what I am doing wrong?
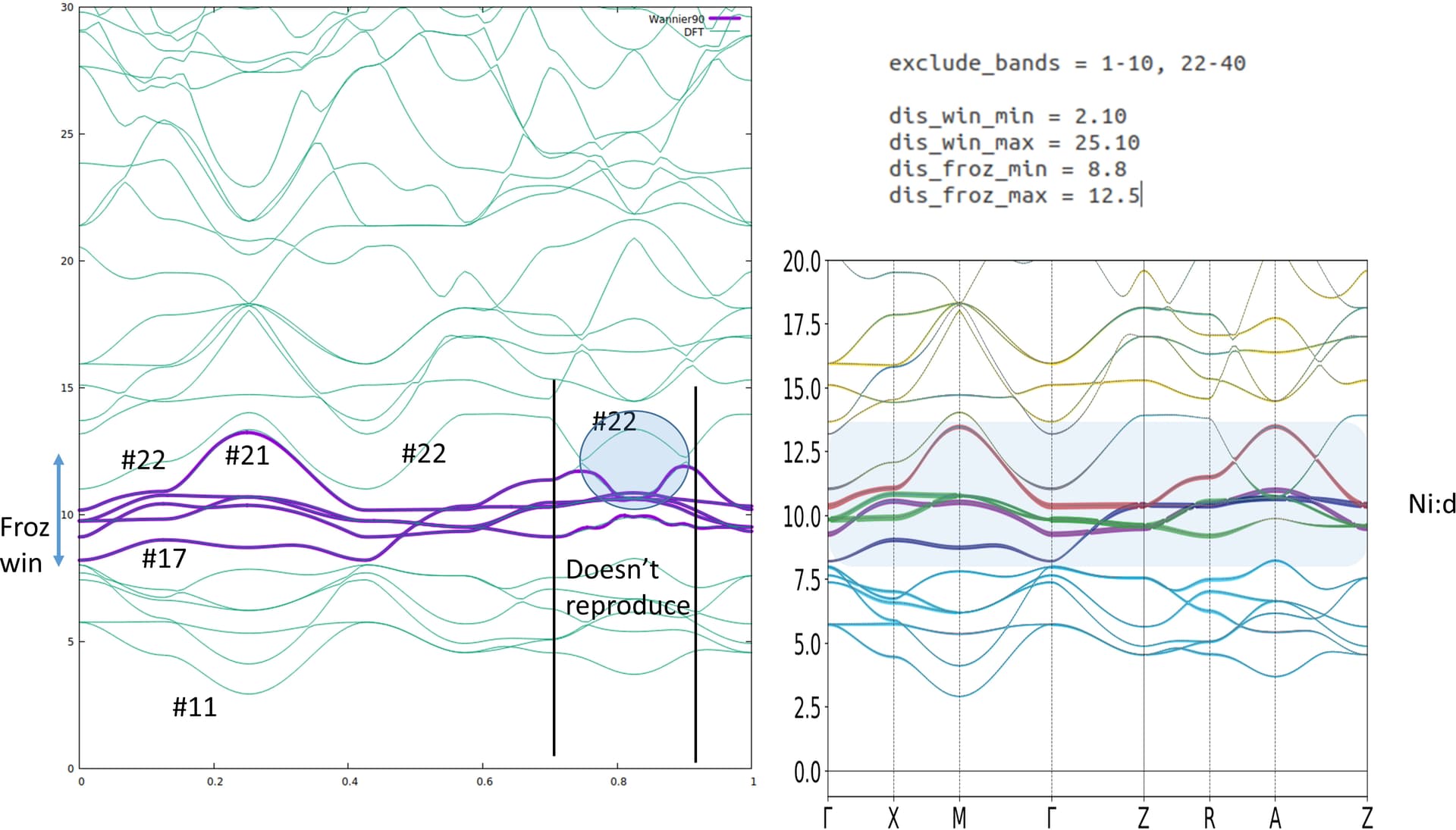
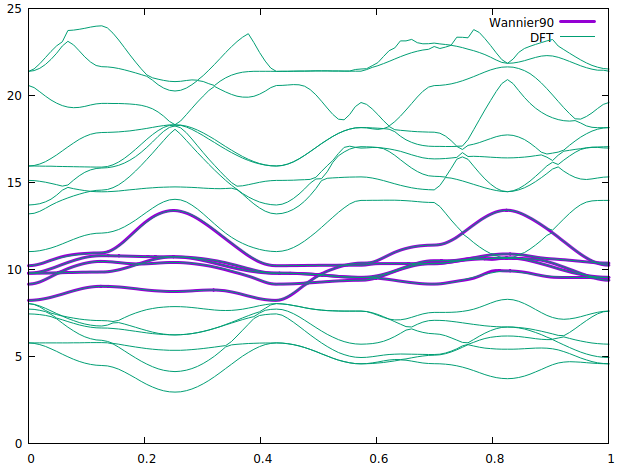
In the first figure, right side shows DFT bands where ni-3d bands are in the shaded region. The top red one is the dx2-y2. In the left one we have wannier90 bands (purple). It reproduces all most every part of ni:d bands except in the marked circle. This part should be inverted and goes to band #22. The energy window also attached in the picture. I excluded the band #22.
When I take the band #22 into account as you can see in the 2nd picture, the wannier dx2-y2 band is completely gone. This could be due to the max frozen window which is at 10.5. However, I can not increase the Froz_max as it gives an error saying " more states in the frozen window than the target WFs"
Any help to solve this issue please.
Regards,
Rajesh
Hi Rajesh,
When the band #22 is excluded, wannier90 will try to get the best fit for the band INSIDE the frozen window, which gives exactly what you get. If you want to avoid the band in the circle, this part shouldn’t be included in the frozen window, and #22 should be included in the disentanglement window. So the top of the frozen window should be lowered. However, this does not guarantee that the d-bands you’re looking for will be generated. You can try tuning the energy windows and rely on luck…
A better strategy is probably to increase the number of projectors so that more states can be included in the frozen window. In the RNiO2 structures, the usual strategy is to add a s-like orbital in the R site. Then you can shift the top of the frozen window up to make sure the d-band in the circle is included. This actually gives a good description of the physics that there is some self-doping effect due to the A-site.
Best regards,
Xu He
Dear Xu,
There is no significant contribution from s-like orbital in the R-site if we see the DOS. However, in the literature people say interstitial-s orbital of apical oxygen which sits in the (0,0,0.5) type positions. In our calculation there is no such position in the atom block. Therefore, I don’t know how to include such s-orbital in the wannier90 input file.
It works anyway when I take take “num_wann=6” and define only 5-Ni:d bands
begin projections
Ni:d
end projections
It seems it needs one more WF, therefore, my question is that is it possible to have num_wann > number of projections? If yes, then question would be which wannier orbital is taken by wannier90 for band#22 since I have not specified in the projection block?
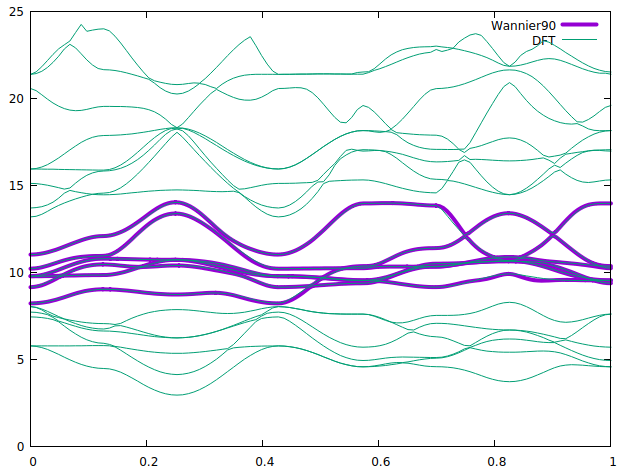
Hi @mailhexu,
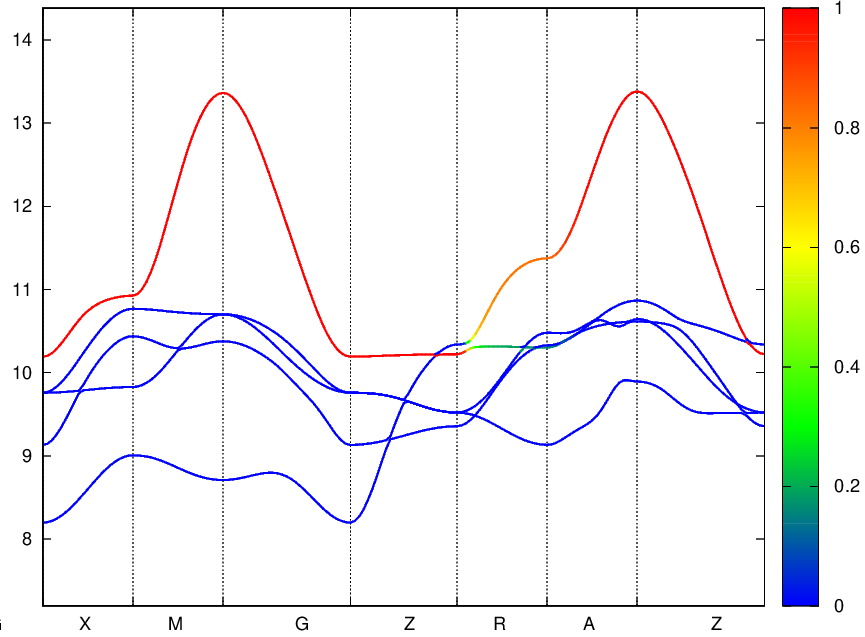
I finally got it using just 5-Ni:d projections. I just had to lower the Froz_Max including band #22. Here is the plot.
BTW, is there any way to extract the orbital resolved weight from wannier bands ?
Best!
Rajesh
Dear Rajesh,
To get orbital character, you need to compute the overlap between the band state and the orbital state. I’m not aware of an automatic tool, but you can do it by taking the Hamiltonian in the orbital basis at every k-point, computing the different eigenvectors/eigenvalues and compute the overlap at every k-point.
Best,
Olivier
Dear @gingras.ol,
I think with wannier90 it can be done using the “bands_plot_project” input in the .win file and it gives nice result!
Here is an example below.
Best!
Rajesh
Oh right! Very nice indeed
Olivier